SUPPLEMENTARY APPENDIX Exome Sequencing Reveals Heterogeneous Clonal Dynamics in Donor Cell Myeloid Neoplasms After Stem Cell Transplantation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands
The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands Clare Louise Wishart Submitted in accordance with the requirements for the degree of Doctor of Philosophy of Science University of Leeds School of Biomedical Sciences Faculty of Biological Sciences September 2013 I Intellectual Property and Publication Statements The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Clare Louise Wishart to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2013 The University of Leeds and Clare Louise Wishart. II Acknowledgments Firstly I would like to offer my sincerest thanks and gratitude to my supervisor, Dr. Dan Donnelly, who has been nothing but encouraging and engaging from day one. I have thoroughly enjoyed every moment of working alongside him and learning from his guidance and wisdom. My thanks go to my academic assessor Professor Paul Milner whom I have known for several years, and during my time at the University of Leeds he has offered me invaluable advice and inspiration. Additionally I would like to thank my academic project advisor Dr. Michael Harrison for his friendship, help and advice. I would like to thank Dr. Rosalind Mann and Dr. Elsayed Nasr for welcoming me into the lab as a new PhD student and sharing their experimental techniques with me, these techniques have helped me no end in my time as a research student. -

Downloaded from the National Database for Autism Research (NDAR)
International Journal of Molecular Sciences Article Phenotypic Subtyping and Re-Analysis of Existing Methylation Data from Autistic Probands in Simplex Families Reveal ASD Subtype-Associated Differentially Methylated Genes and Biological Functions Elizabeth C. Lee y and Valerie W. Hu * Department of Biochemistry and Molecular Medicine, The George Washington University, School of Medicine and Health Sciences, Washington, DC 20037, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-202-994-8431 Current address: W. Harry Feinstone Department of Molecular Microbiology and Immunology, y Johns Hopkins Bloomberg School of Public Health, Baltimore, MD 21205, USA. Received: 25 August 2020; Accepted: 17 September 2020; Published: 19 September 2020 Abstract: Autism spectrum disorder (ASD) describes a group of neurodevelopmental disorders with core deficits in social communication and manifestation of restricted, repetitive, and stereotyped behaviors. Despite the core symptomatology, ASD is extremely heterogeneous with respect to the severity of symptoms and behaviors. This heterogeneity presents an inherent challenge to all large-scale genome-wide omics analyses. In the present study, we address this heterogeneity by stratifying ASD probands from simplex families according to the severity of behavioral scores on the Autism Diagnostic Interview-Revised diagnostic instrument, followed by re-analysis of existing DNA methylation data from individuals in three ASD subphenotypes in comparison to that of their respective unaffected siblings. We demonstrate that subphenotyping of cases enables the identification of over 1.6 times the number of statistically significant differentially methylated regions (DMR) and DMR-associated genes (DAGs) between cases and controls, compared to that identified when all cases are combined. Our analyses also reveal ASD-related neurological functions and comorbidities that are enriched among DAGs in each phenotypic subgroup but not in the combined case group. -
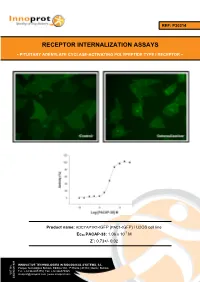
Receptor Internalization Assays
REF: P30214 RECEPTOR INTERNALIZATION ASSAYS - PITUITARY ADENYLATE CYCLASE-ACTIVATING POLYPEPTIDE TYPE I RECEPTOR - Product name: ADCYAP1R1-tGFP (PAC1-tGFP) / U2OS cell line -7 Ec50 PACAP-38: 1.06 x 10 M Z´: 0.73+/- 0.02 INNOVATIVE TECHNOLOGIES IN BIOLOGICAL SYSTEMS, S.L. Parque Tecnológico Bizkaia, Edifício 502, 1ª Planta | 48160 | Derio | Bizkaia Tel.: +34 944005355 | Fax: +34 946579925 VAT No. [email protected] | www.innoprot.com ESB95481909 Product Name: ADCYAP1R1-tGFP_U2OS Reference: P30214 Rep. Official Full Name: Pituitary adenylate cyclase- activating polypeptide type I receptor DNA Accession Number: Gene Bank AY366498 Host Cell: U2OS References: P30214: 2 vials of 3 x 106 proliferative cells P30214-DA: 1 vial of 2 x 106 division-arrested cells Storage: Liquid Nitrogen Assay Briefly description About ADCYAP1R1 Each vial of ADCYAP1R1 Internalization Assay Pituitary adenylate cyclase-activating Cell Line contains U2OS cells stably expressing polypeptide type I receptor, also known as human Pituitary adenylate cyclase-activating PAC1 is a protein that in humans is encoded by polypeptide type I receptor tagged in the N- the ADCYAP1R1 gene. ADCYAP1R1 is a terminus with tGFP protein. membrane-associated protein and shares significant homology with members of the Innoprot’s ADCYAP1R1-tGFP Internalization glucagon/secretin receptor family. This receptor Assay Cell Line has been designed to assay binds pituitary adenylate cyclase activating potential agonists/ antagonists against peptide (PACAP) mediating several biological ADCYAP1R1, modulating its activation and the activities and it is positively coupled to following redistribution process inside the cells. adenylate cyclase. This cell line will allow the image analysis of the stimuli induced by the compounds. -

Beta-Arrestin-Mediated Signaling in the Heart
SPECIAL ARTICLE Circ J 2008; 72: 1725–1729 Beta-Arrestin-Mediated Signaling in the Heart Priyesh A. Patel, BS; Douglas G. Tilley, PhD*; Howard A. Rockman, MD*,** Beta-arrestin is a multifunctional adapter protein well known for its role in G-protein-coupled receptor (GPCR) desensitization. Exciting new evidence indicates thatβ-arrestin is also a signaling molecule capable of initiating its own G-protein-independent signaling at GPCRs. One of the best-studiedβ-arrestin signaling pathways is the one involvingβ-arrestin-dependent activation of a mitogen-activated protein kinase cascade, the extracellular regulated kinase (ERK). ERK signaling, which is classically activated by agonist stimulation of the epidermal growth factor receptor (EGFR), can be activated by a number of GPCRs in aβ-arrestin-dependent manner. Recent work in animal models of heart failure suggests thatβ-arrestin-dependent activation of EGFR/ERK signaling by theβ-1-adrenergic receptor, and possibly the angiotensin II Type 1A receptor, are cardioprotective. Hence, a new model of signaling at cardiac GPCRs has emerged and implicates classical G-protein-mediated signaling with promoting harmful remodeling in heart failure, while concurrently linkingβ-arrestin-dependent, G-protein-inde- pendent signaling with cardioprotective effects. Based on this paradigm, a new class of drugs could be identified, termed “biased ligands”, which simultaneously block harmful G-protein signaling, while also promoting cardio- protectiveβ-arrestin-dependent signaling, leading to a potential breakthrough -

BMC Evolutionary Biology Biomed Central
BMC Evolutionary Biology BioMed Central Research article Open Access On the origins of arrestin and rhodopsin Carlos E Alvarez1,2,3 Address: 1Center for Molecular and Human Genetics, The Research Institute at Nationwide Children's Hospital, Columbus, OH, 43205, USA, 2Department of Pediatrics, The Ohio State University College of Medicine, Columbus, OH, 43210, USA and 3Novartis Institutes of BioMedical Research, CH-4002 Basel, Switzerland Email: Carlos E Alvarez - [email protected] Published: 29 July 2008 Received: 11 January 2008 Accepted: 29 July 2008 BMC Evolutionary Biology 2008, 8:222 doi:10.1186/1471-2148-8-222 This article is available from: http://www.biomedcentral.com/1471-2148/8/222 © 2008 Alvarez; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: G protein coupled receptors (GPCRs) are the most numerous proteins in mammalian genomes, and the most common targets of clinical drugs. However, their evolution remains enigmatic. GPCRs are intimately associated with trimeric G proteins, G protein receptor kinases, and arrestins. We conducted phylogenetic studies to reconstruct the history of arrestins. Those findings, in turn, led us to investigate the origin of the photosensory GPCR rhodopsin. Results: We found that the arrestin clan is comprised of the Spo0M protein family in archaea and bacteria, and the arrestin and Vps26 families in eukaryotes. The previously known animal arrestins are members of the visual/beta subfamily, which branched from the founding "alpha" arrestins relatively recently. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Supporting Online Material
1 2 3 4 5 6 7 Supplementary Information for 8 9 Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic 10 retinopathy 11 12 *Samuel A. Mills, *Andrew I. Jobling, *Michael A. Dixon, Bang V. Bui, Kirstan A. Vessey, Joanna A. Phipps, 13 Ursula Greferath, Gene Venables, Vickie H.Y. Wong, Connie H.Y. Wong, Zheng He, Flora Hui, James C. 14 Young, Josh Tonc, Elena Ivanova, Botir T. Sagdullaev, Erica L. Fletcher 15 * Joint first authors 16 17 Corresponding author: 18 Prof. Erica L. Fletcher. Department of Anatomy & Neuroscience. The University of Melbourne, Grattan St, 19 Parkville 3010, Victoria, Australia. 20 Email: [email protected] ; Tel: +61-3-8344-3218; Fax: +61-3-9347-5219 21 22 This PDF file includes: 23 24 Supplementary text 25 Figures S1 to S10 26 Tables S1 to S7 27 Legends for Movies S1 to S2 28 SI References 29 30 Other supplementary materials for this manuscript include the following: 31 32 Movies S1 to S2 33 34 35 36 1 1 Supplementary Information Text 2 Materials and Methods 3 Microglial process movement on retinal vessels 4 Dark agouti rats were anaesthetized, injected intraperitoneally with rhodamine B (Sigma-Aldrich) to label blood 5 vessels and retinal explants established as described in the main text. Retinal microglia were labelled with Iba-1 6 and imaging performed on an inverted confocal microscope (Leica SP5). Baseline images were taken for 10 7 minutes, followed by the addition of PBS (10 minutes) and then either fractalkine or fractalkine + candesartan 8 (10 minutes) using concentrations outlined in the main text. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Sodium Pumps Mediate Activity-Dependent Changes in Mammalian Motor Networks
906 • The Journal of Neuroscience, January 25, 2017 • 37(4):906–921 Systems/Circuits Sodium Pumps Mediate Activity-Dependent Changes in Mammalian Motor Networks X Laurence D. Picton, XFilipe Nascimento, XMatthew J. Broadhead, XKeith T. Sillar, and XGareth B. Miles School of Psychology and Neuroscience, University of St Andrews, St Andrews KY16 9JP, United Kingdom Ubiquitously expressed sodium pumps are best known for maintaining the ionic gradients and resting membrane potential required for generating action potentials. However, activity- and state-dependent changes in pump activity can also influence neuronal firing and regulate rhythmic network output. Here we demonstrate that changes in sodium pump activity regulate locomotor networks in the spinal cord of neonatal mice. The sodium pump inhibitor, ouabain, increased the frequency and decreased the amplitude of drug-induced locomotor bursting, effects that were dependent on the presence of the neuromodulator dopamine. Conversely, activating the pump with the sodium ionophore monensin decreased burst frequency. When more “natural” locomotor output was evoked using dorsal-root stimulation, ouabain increased burst frequency and extended locomotor episode duration, whereas monensin slowed and shortened episodes. Decreasing the time between dorsal-root stimulation, and therefore interepisode interval, also shortened and slowed activity, suggesting that pump activity encodes information about past network output and contributes to feedforward control of subsequent locomotor bouts. Using whole-cell patch-clamp recordings from spinal motoneurons and interneurons, we describe a long-duration (ϳ60 s), activity-dependent, TTX- and ouabain-sensitive, hyperpolarization (ϳ5 mV), which is mediated by spike-dependent increases in pump activity. The duration of this dynamic pump potential is enhanced by dopamine. -

A SARS-Cov-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing
A SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets and Potential Drug-Repurposing Supplementary Information Supplementary Discussion All SARS-CoV-2 protein and gene functions described in the subnetwork appendices, including the text below and the text found in the individual bait subnetworks, are based on the functions of homologous genes from other coronavirus species. These are mainly from SARS-CoV and MERS-CoV, but when available and applicable other related viruses were used to provide insight into function. The SARS-CoV-2 proteins and genes listed here were designed and researched based on the gene alignments provided by Chan et. al. 1 2020 . Though we are reasonably sure the genes here are well annotated, we want to note that not every protein has been verified to be expressed or functional during SARS-CoV-2 infections, either in vitro or in vivo. In an effort to be as comprehensive and transparent as possible, we are reporting the sub-networks of these functionally unverified proteins along with the other SARS-CoV-2 proteins. In such cases, we have made notes within the text below, and on the corresponding subnetwork figures, and would advise that more caution be taken when examining these proteins and their molecular interactions. Due to practical limits in our sample preparation and data collection process, we were unable to generate data for proteins corresponding to Nsp3, Orf7b, and Nsp16. Therefore these three genes have been left out of the following literature review of the SARS-CoV-2 proteins and the protein-protein interactions (PPIs) identified in this study. -

Intrinsic Disorder in PTEN and Its Interactome Confers Structural Plasticity and 63
OPEN Intrinsic Disorder in PTEN and its SUBJECT AREAS: Interactome Confers Structural Plasticity CELLULAR SIGNALLING NETWORKS and Functional Versatility CANCER GENOMICS Prerna Malaney1*, Ravi Ramesh Pathak1*, Bin Xue3, Vladimir N. Uversky3,4,5 & Vrushank Dave´1,2 SYSTEMS ANALYSIS GENE REGULATORY NETWORKS 1Morsani College of Medicine, Department of Pathology and Cell Biology, 2Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, 3Department of Molecular Medicine, 4USF Health Byrd Alzheimer’s Research Institute, Received University of South Florida, Tampa, FL, 33612, USA, 5Institute for Biological Instrumentation, Russian Academy of Sciences, 142290 11 March 2013 Pushchino, Moscow Region, Russia. Accepted 3 June 2013 IDPs, while structurally poor, are functionally rich by virtue of their flexibility and modularity. However, how mutations in IDPs elicit diseases, remain elusive. Herein, we have identified tumor suppressor PTEN as Published an intrinsically disordered protein (IDP) and elucidated the molecular principles by which its intrinsically 20 June 2013 disordered region (IDR) at the carboxyl-terminus (C-tail) executes its functions. Post-translational modifications, conserved eukaryotic linear motifs and molecular recognition features present in the C-tail IDR enhance PTEN’s protein-protein interactions that are required for its myriad cellular functions. PTEN primary and secondary interactomes are also enriched in IDPs, most being cancer related, revealing that Correspondence and PTEN functions emanate from and are nucleated by the C-tail IDR, which form pliable network-hubs. requests for materials Together, PTEN higher order functional networks operate via multiple IDP-IDP interactions facilitated by should be addressed to its C-tail IDR. Targeting PTEN IDR and its interaction hubs emerges as a new paradigm for treatment of V.D.