The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Strategies to Increase ß-Cell Mass Expansion
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Strategies to increase -cell mass expansion Drynda, Robert Lech Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 Strategies to increase β-cell mass expansion A thesis submitted by Robert Drynda For the degree of Doctor of Philosophy from King’s College London Diabetes Research Group Division of Diabetes & Nutritional Sciences Faculty of Life Sciences & Medicine King’s College London 2017 Table of contents Table of contents ................................................................................................. -

Exenatide QW: a New Treatment Option for Type 2 Diabetes Offering Ease of Use, Improved Efficacy, and Reduced Side Effects
FEATURE ARTICLE Commentary: Exenatide QW: A New Treatment Option for Type 2 Diabetes Offering Ease of Use, Improved Efficacy, and Reduced Side Effects Charles F. Shaefer, Jr., MD Editor’s note: Once-weekly exenatide, (also called incretin mimetics) cur- with GLP-1 receptor agonists is the which has recently been approved for rently in use.3,4 only form of anti-diabetes therapy patients with type 2 diabetes, has great Validation of the acceptance offering proven weight loss. Finally, potential as a new diabetes therapy in clinical practice of GLP-1 GLP-1 receptor agonist therapy is in the primary care setting. This receptor agonists can be found in one of the recommended treatment commentary and the feature article that the recently released American strategies offering a low incidence of follows it (p. 95) offer an overview of Diabetes Association (ADA)/ hypoglycemia, an important aspect this new therapeutic tool and important European Association for the Study of diabetes therapy learned from the insights about its clinical utility. In the of Diabetes (EASD) position state- Action to Control Cardiovascular interest of transparency, however, we ment on the management of type 2 Risk in Diabetes trial.6 want to point out that the authors of diabetes, which placed these drugs As clinicians consider what to do both articles are affiliated with Amylin alongside older, well-accepted agents when metformin and lifestyle change Pharmaceuticals, which manufactures such as sulfonylureas (SUs) and do not adequately control a patient’s exenatide QW and markets it under the thiozolidinediones (TZDs) as a sec- A1C, they frequently ask, “incretin 5 trade name Bydureon. -

GLP-1 Receptor Agonists
Cognitive Vitality Reports® are reports written by neuroscientists at the Alzheimer’s Drug Discovery Foundation (ADDF). These scientific reports include analysis of drugs, drugs-in- development, drug targets, supplements, nutraceuticals, food/drink, non-pharmacologic interventions, and risk factors. Neuroscientists evaluate the potential benefit (or harm) for brain health, as well as for age-related health concerns that can affect brain health (e.g., cardiovascular diseases, cancers, diabetes/metabolic syndrome). In addition, these reports include evaluation of safety data, from clinical trials if available, and from preclinical models. GLP-1 Receptor Agonists Evidence Summary GLP-1 agonists are beneficial for patients with type 2 diabetes and obesity. Some evidence suggests benefits for Alzheimer’s disease. It is unclear whether it is beneficial for individuals without underlying metabolic disease. Semaglutide seems to be most effective for metabolic dysfunction, though liraglutide has more preclinical data for Alzheimer’s disease. Neuroprotective Benefit: Evidence from many preclinical studies and a pilot biomarker study suggest some neuroprotective benefits with GLP-1 agonists. However, whether they may be beneficial for everyone or only a subset of individuals (e.g. diabetics) is unclear. Aging and related health concerns: GLP-1 agonists are beneficial for treating diabetes and cardiovascular complications relating to diabetes. It is not clear whether they have beneficial effects in otherwise healthy individuals. Safety: GLP-1 agonists are generally safe for most people with minor side effects. However, long-term side effects are not known. 1 Availability: Available Dose: Varies - see Chemical formula: C172H265N43O51 (Liraglutide) as a prescription chart at the end of MW: 3751.262 g/mol medicine. -

Review of Current Real-World Experience with Teriparatide As Treatment of Osteoporosis in Different Patient Groups
Journal of Clinical Medicine Review Review of Current Real-World Experience with Teriparatide as Treatment of Osteoporosis in Different Patient Groups Barbara Hauser 1,2,* , Nerea Alonso 2 and Philip L Riches 1,2 1 Rheumatic Disease Unit, Western General Hospital, NHS Lothian, Edinburgh EH4 2XU, UK; [email protected] 2 Rheumatology and Bone Disease Unit, Centre for Genomic and Experimental Medicine, MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh EH4 2XU, UK; [email protected] * Correspondence: [email protected] Abstract: Teriparatide has proven effective in reducing both vertebral and non-vertebral fractures in clinical trials of post-menopausal and glucocorticoid-induced osteoporosis. Widespread adoption of Teriparatide over the last two decades means that there is now substantial experience of its use in routine clinical practice, which is summarized in this paper. Extensive real-world experience of Teriparatide in post-menopausal osteoporosis confirms the fracture and bone density benefits seen in clinical trials, with similar outcomes identified also in male and glucocorticoid-induced osteoporosis. Conversely, very limited experience has been reported in pre-menopausal osteoporosis or in the use of Teriparatide in combination with other therapies. Surveillance studies have identified no safety signals relating to the possible association of Teriparatide with osteosarcoma. We also review the evidence for predicting response to Teriparatide in order to inform the debate on where best to use Teriparatide in an increasingly crowded therapeutic landscape. Citation: Hauser, B.; Alonso, N.; Riches, P.L. Review of Current Keywords: Teriparatide; anabolic treatment; osteoporosis; fracture Real-World Experience with Teriparatide as Treatment of Osteoporosis in Different Patient Groups. -

The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer's Disease
Edith Cowan University Research Online ECU Publications Post 2013 3-31-2016 The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer's Disease Ian James Martins Edith Cowan University Follow this and additional works at: https://ro.ecu.edu.au/ecuworkspost2013 Part of the Medicine and Health Sciences Commons 10.3390/proteomes4020014 Martins, I. J. (2016). The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease. Proteomes, 4(2), 14.Available here This Journal Article is posted at Research Online. https://ro.ecu.edu.au/ecuworkspost2013/2456 proteomes Article The Role of Clinical Proteomics, Lipidomics, and Genomics in the Diagnosis of Alzheimer’s Disease Ian James Martins School of Medical Sciences, Edith Cowan University, 270 Joondalup Drive, Joondalup 6027, Australia; [email protected]; Tel.: +61-8-6304-2574 Academic Editors: Edwin Lasonder and Jacek R. Wisniewski Received: 5 February 2016; Accepted: 28 March 2016; Published: 31 March 2016 Abstract: The early diagnosis of Alzheimer’s disease (AD) has become important to the reversal and treatment of neurodegeneration, which may be relevant to premature brain aging that is associated with chronic disease progression. Clinical proteomics allows the detection of various proteins in fluids such as the urine, plasma, and cerebrospinal fluid for the diagnosis of AD. Interest in lipidomics has accelerated with plasma testing for various lipid biomarkers that may with clinical proteomics provide a more reproducible diagnosis for early brain aging that is connected to other chronic diseases. The combination of proteomics with lipidomics may decrease the biological variability between studies and provide reproducible results that detect a community’s susceptibility to AD. -

Cloning, Characterization, and Expression of a Human Calcitonin Receptor from an Ovarian Carcinoma Cell Line
Cloning, characterization, and expression of a human calcitonin receptor from an ovarian carcinoma cell line. A H Gorn, … , S M Krane, S R Goldring J Clin Invest. 1992;90(5):1726-1735. https://doi.org/10.1172/JCI116046. Research Article A human ovarian small cell carcinoma line (BIN-67) expresses abundant calcitonin (CT) receptors (CTR) (143,000 per cell) that are coupled, to adenylate cyclase. The dissociation constants (Kd) for the CTRs on these BIN-67 cells is approximately 0.42 nM for salmon CT and approximately 4.6 nM for human CT. To clone a human CTR (hCTR), a BIN-67 cDNA library was screened using a cDNA probe from a porcine renal CTR (pCTR) that we recently cloned. One positive clone of 3,588 bp was identified. Transfection of this cDNA into COS cells resulted in expression of receptors with high affinity for salmon CT (Kd = approximately 0.44 nM) and for human CT (Kd = approximately 5.4 nM). The expressed hCTR was coupled to adenylate cyclase. Northern analysis with the hCTR cDNA probe indicated a single transcript of approximately 4.2 kb. The cloned cDNA encodes a putative peptide of 490 amino acids with seven potential transmembrane domains. The amino acid sequence of the hCTR is 73% identical to the pCTR, although the hCTR contains an insert of 16 amino acids between transmembrane domain I and II. The structural differences may account for observed differences in binding affinity between the porcine renal and human ovarian CTRs. The CTRs are closely related to the receptors for parathyroid hormone-parathyroid hormone-related peptide and secretin; these receptors comprise a […] Find the latest version: https://jci.me/116046/pdf Cloning, Characterization, and Expression of a Human Calcitonin Receptor from an Ovarian Carcinoma Cell Line Alan H. -
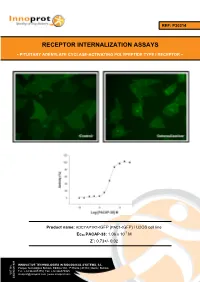
Receptor Internalization Assays
REF: P30214 RECEPTOR INTERNALIZATION ASSAYS - PITUITARY ADENYLATE CYCLASE-ACTIVATING POLYPEPTIDE TYPE I RECEPTOR - Product name: ADCYAP1R1-tGFP (PAC1-tGFP) / U2OS cell line -7 Ec50 PACAP-38: 1.06 x 10 M Z´: 0.73+/- 0.02 INNOVATIVE TECHNOLOGIES IN BIOLOGICAL SYSTEMS, S.L. Parque Tecnológico Bizkaia, Edifício 502, 1ª Planta | 48160 | Derio | Bizkaia Tel.: +34 944005355 | Fax: +34 946579925 VAT No. [email protected] | www.innoprot.com ESB95481909 Product Name: ADCYAP1R1-tGFP_U2OS Reference: P30214 Rep. Official Full Name: Pituitary adenylate cyclase- activating polypeptide type I receptor DNA Accession Number: Gene Bank AY366498 Host Cell: U2OS References: P30214: 2 vials of 3 x 106 proliferative cells P30214-DA: 1 vial of 2 x 106 division-arrested cells Storage: Liquid Nitrogen Assay Briefly description About ADCYAP1R1 Each vial of ADCYAP1R1 Internalization Assay Pituitary adenylate cyclase-activating Cell Line contains U2OS cells stably expressing polypeptide type I receptor, also known as human Pituitary adenylate cyclase-activating PAC1 is a protein that in humans is encoded by polypeptide type I receptor tagged in the N- the ADCYAP1R1 gene. ADCYAP1R1 is a terminus with tGFP protein. membrane-associated protein and shares significant homology with members of the Innoprot’s ADCYAP1R1-tGFP Internalization glucagon/secretin receptor family. This receptor Assay Cell Line has been designed to assay binds pituitary adenylate cyclase activating potential agonists/ antagonists against peptide (PACAP) mediating several biological ADCYAP1R1, modulating its activation and the activities and it is positively coupled to following redistribution process inside the cells. adenylate cyclase. This cell line will allow the image analysis of the stimuli induced by the compounds. -

List of Approved Ndas for Biological Products That Were Deemed to Be Blas on March 23, 2020
List of Approved NDAs for Biological Products That Were Deemed to be BLAs on March 23, 2020 On March 23, 2020, an approved application for a biological product under section 505 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) was deemed to be a license for the biological product under section 351 of the Public Health Service Act (PHS Act) (see section 7002(e)(4)(A) of the Biologics Price Competition and Innovation Act of 2009). To enhance transparency and facilitate planning for the March 23, 2020, transition date, FDA compiled a preliminary list of approved applications for biological products under the FD&C Act that were listed in FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book) and that would be affected by this transition provision. FDA posted this list on the FDA website in December 2018, and periodically updated this list before the March 23, 2020, transition date. The September 2019 update to this preliminary list added certain administratively closed applications related to approved applications for biological products that were on the December 2018 version of this list. The January 2020 update to the preliminary list reflected a change to the definition of “biological product” made by the Further Consolidated Appropriations Act, 2020, which was enacted on December 20, 2019. Section 605 of this Act further amended the definition of a “biological product” in section 351(i) of the PHS Act to remove the parenthetical “(except any chemically synthesized polypeptide)” from the statutory category of “protein.” FDA has provided below a list of each approved application for a biological product under the FD&C Act that was deemed to be a license (i.e., an approved biologics license application (BLA)) for the biological product on March 23, 2020. -

Corticorelin Acetate, a Synthetic Corticotropin-Releasing Factor with Preclinical Antitumor Activity, Alone and with Bevacizumab, Against Human Brain Tumor Models
ANTICANCER RESEARCH 30: 5037-5042 (2010) Corticorelin Acetate, a Synthetic Corticotropin-releasing Factor with Preclinical Antitumor Activity, alone and with Bevacizumab, against Human Brain Tumor Models IDOIA GAMEZ1, ROBERT P. RYAN1 and STEPHEN T. KEIR2 1Celtic Pharmaceutical Development Services America, Inc., New York, NY 10022, U.S.A.; 2The Preston Robert Tisch Brain Tumor Center, Duke University, Durham, NC 27710, U.S.A. Abstract. Background: Corticorelin acetate (CrA) is a brain tumors. The cause of PBE is believed to result in part synthetic form of corticotropin-releasing factor that is from the leakage of edematous brain tumor fluid from currently undergoing clinical trials in the treatment of abnormal tumor vasculature into the surrounding tissue (1, peritumoral brain edema (PBE). This study preclinically 2). The mechanism(s) of action by which CrA exerts its investigated its potential as an antitumor agent against beneficial effect in the therapy of PBE has not been human brain tumor xenografts. Materials and Methods: The elucidated, but two factors appear to be relevant: decreased in vivo efficacy of CrA as a single agent and in combination vascular leakage and preserved integrity of the endothelial with the antiangiogenic agent, bevacizumab, was examined cells, which ultimately helps to maintain the blood-brain in three different patient-derived human brain tumor barrier (3, 4). xenografts implanted orthotopically (intracranially) or CrA appears to mediate its activity through two subtypes subcutaneously in athymic mice. Results: CrA significantly of G-protein coupled receptors: CRF receptor-1 (CRFR1) increased the lifespan of mice implanted orthotopically with and CRF receptor-2 (CRFR2) (5). These CRFRs are widely two different pediatric brain tumor xenograft models. -

(Pram) and Insulin A21G Improves Post-Prandial Glucose Vs Novolog
ADO09, A Co-Formulation Of Pramlintide (Pram) and Insulin A21G improves Post-Prandial Glucose Vs Novolog® in Type 1 Diabetes (T1DM) G.Meiffren¹, G.Andersen², R.Eloy¹, C.Seroussi¹, C.Mégret¹, S.Famulla², Y.-P Chan¹, M.Gaudier¹, O.Soula¹, J.H. DeVries²,T.Heise² (1 Adocia, Lyon, France ; 2 Profil, Neuss, Germany) Introduction & Background Overall safety Outpatient period results - CGM metrics o ADO09 (M1Pram) is a co-formulation of pramlintide and insulin A21G o Both treatments were well tolerated without any treatment-related serious adverse events o Most of the CGM metrics (TiR [70-180], TiR [80-140], mean blood glucose per day), were significantly improved developed to leverage the beneficial effects of pramlintide on post-prandial (Table 2). As expected M1Pram had numerically more, mostly gastrointestinal adverse events with M1Pram (Table 4). Postprandial and mean 24-hour glucose profiles were improved with M1Pram (Fig. 3) glucose without additional injections than insulin aspart Table 4: CGM metrics, all days. Significant differences are marked in bold Objective and design o No severe hypoglycemia were seen, slightly more hypoglycemic events occurred with M1Pram Ratio of LSMean* o To compare the effect of M1Pram and insulin aspart (Novolog®, Novo than with aspart (Table 3) Difference Parameter Treatment LS Mean M1Pram / Aspart P-value Nordisk) on post-prandial glucose control, glycemic control assessed by Table 2: Incidence of adverse events throughout the trial (M1Pram-Aspart) (95% CI) CGM and safety/tolerability M1Pram Aspart M1Pram -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Gattex Teduglutide Rdna Origin
Subject: Gattex (teduglutide [rDNA origin]) Original Effective Date: 5/30/2014 Policy Number: MCP-176 Revision Date(s): Review Date(s): 12/16/15; 9/15/2016; 6/22/2017 DISCLAIMER This Medical Policy is intended to facilitate the Utilization Management process. It expresses Molina's determination as to whether certain services or supplies are medically necessary, experimental, investigational, or cosmetic for purposes of determining appropriateness of payment. The conclusion that a particular service or supply is medically necessary does not constitute a representation or warranty that this service or supply is covered (i.e., will be paid for by Molina) for a particular member. The member's benefit plan determines coverage. Each benefit plan defines which services are covered, which are excluded, and which are subject to dollar caps or other limits. Members and their providers will need to consult the member's benefit plan to determine if there are any exclusion(s) or other benefit limitations applicable to this service or supply. If there is a discrepancy between this policy and a member's plan of benefits, the benefits plan will govern. In addition, coverage may be mandated by applicable legal requirements of a State, the Federal government or CMS for Medicare and Medicaid members. CMS's Coverage Database can be found on the CMS website. The coverage directive(s) and criteria from an existing National Coverage Determination (NCD) or Local Coverage Determination (LCD) will supersede the contents of this Molina medical coverage policy (MCP) document and provide the directive for all Medicare members. FDA INDICATIONS � ∑ Short bowel syndrome: For the treatment of adult patients with short bowel syndrome who are dependent on parenteral support.