Modeling and Molecular Dynamics Indicate That Snake Venom Phospholipase B-Like Enzymes Are Ntn-Hydrolases T
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Materials Supplemental Table 1
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2016 Supplemental Materials Supplemental Table 1. The differentially expressed proteins from rat pancreas identified by proteomics (SAP vs. SO) No. Protein name Gene name ratio P value 1 Metallothionein Mt1m 3.35 6.34E-07 2 Neutrophil antibiotic peptide NP-2 Defa 3.3 8.39E-07 3 Ilf2 protein Ilf2 3.18 1.75E-06 4 Numb isoform o/o rCG 3.12 2.73E-06 5 Lysozyme Lyz2 3.01 5.63E-06 6 Glucagon Gcg 2.89 1.17E-05 7 Serine protease HTRA1 Htra1 2.75 2.97E-05 8 Alpha 2 macroglobulin cardiac isoform (Fragment) 2.75 2.97E-05 9 Myosin IF (Predicted) Myo1f 2.65 5.53E-05 10 Neuroendocrine secretory protein 55 Gnas 2.61 7.60E-05 11 Matrix metallopeptidase 8 Mmp8 2.57 9.47E-05 12 Protein Tnks1bp1 Tnks1bp1 2.53 1.22E-04 13 Alpha-parvin Parva 2.47 1.78E-04 14 C4b-binding protein alpha chain C4bpa 2.42 2.53E-04 15 Protein KTI12 homolog Kti12 2.41 2.74E-04 16 Protein Rab11fip5 Rab11fip5 2.41 2.84E-04 17 Protein Mcpt1l3 Mcpt1l3 2.33 4.43E-04 18 Phospholipase B-like 1 Plbd1 2.33 4.76E-04 Aldehyde dehydrogenase (NAD), cytosolic 19 2.32 4.93E-04 (Fragments) 20 Protein Dpy19l2 Dpy19l2 2.3 5.68E-04 21 Regenerating islet-derived 3 alpha, isoform CRA_a Reg3a 2.27 6.74E-04 22 60S acidic ribosomal protein P1 Rplp1 2.26 7.22E-04 23 Serum albumin Alb 2.25 7.98E-04 24 Ribonuclease 4 Rnase4 2.24 8.25E-04 25 Cct-5 protein (Fragment) Cct5 2.24 8.52E-04 26 Protein S100-A9 S100a9 2.22 9.71E-04 27 Creatine kinase M-type Ckm 2.21 1.00E-03 28 Protein Larp4b Larp4b 2.18 1.25E-03 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

2013 Kaiser Permanente-Authored Publications Alphabetical by Author
2013 Kaiser Permanente-Authored Publications Alphabetical by Author 1. Abbas MA, Cannom RR, Chiu VY, Burchette RJ, Radner GW, Haigh PI, Etzioni DA Triage of patients with acute diverticulitis: are some inpatients candidates for outpatient treatment? Colorectal Dis. 2013 Apr;15(4):451-7. Southern California 23061533 KP author(s): Abbas, Maher A; Chiu, Vicki Y; Burchette, Raoul J; Radner, Gary W; Haigh, Philip I 2. Abbass MA, Slezak JM, DiFronzo LA Predictors of Early Postoperative Outcomes in 375 Consecutive Hepatectomies: A Single- institution Experience Am Surg. 2013 Oct;79(10):961-7. Southern California 24160779 KP author(s): Abbass, Mohammad Ali; Slezak, Jeffrey M; DiFronzo, Andrew L 3. Abbenhardt C, Poole EM, Kulmacz RJ, Xiao L, Curtin K, Galbraith RL, Duggan D, Hsu L, Makar KW, Caan BJ, Koepl L, Owen RW, Scherer D, Carlson CS, Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) and CCFR, Potter JD, Slattery ML, Ulrich CM Phospholipase A2G1B polymorphisms and risk of colorectal neoplasia Int J Mol Epidemiol Genet. 2013 Sep 12;4(3):140-9. Northern California 24046806 KP author(s): Caan, Bette 4. Aberg JA, Gallant JE, Ghanem KG, Emmanuel P, Zingman BS, Horberg MA Primary Care Guidelines for the Management of Persons Infected With HIV: 2013 Update by the HIV Medicine Association of the Infectious Diseases Society of America Clin Infect Dis. 2013 Nov 13. Mid-Atlantic 24235263 KP author(s): Horberg, Michael 5. Abraham AG, D'Souza G, Jing Y, Gange SJ, Sterling TR, Silverberg MJ, Saag MS, Rourke SB, Rachlis A, Napravnik S, Moore RD, Klein MB, Kitahata MM, Kirk GD, Hogg RS, Hessol NA, Goedert JJ, Gill MJ, Gebo KA, Eron JJ, Engels EA, Dubrow R, Crane HM, Brooks JT, Bosch RJ, Strickler HD, North American AIDS Cohort Collaboration on Research and Design of IeDEA Invasive cervical cancer risk among HIV-infected women: A North American multi-cohort collaboration prospective study J Acquir Immune Defic Syndr. -

World Ratings Draughts
FMJD Rating No 83 January/March 2015 FMJD rating Nr 83 January 2015 email: [email protected] 1 FMJD Rating No 83 January/March 2015 With any question concerning this issue You can contact me directly via email: [email protected] 2 FMJD Rating No 83 January/March 2015 1. Rating - open Name Nationality Title Rating +/- 1 = shvartsman alexander Russia gmi 2425 -19 2 = georgiev alexander Russia gmi 2421 -15 3 +5 chizhov alexey Russia gmi 2409 +18 4 = getmanski alexander Russia gmi 2404 +4 5 +1 boomstra roel Netherlands gmi 2399 +4 6 -3 baliakin alexander Netherlands gmi 2396 -11 7 = gantvarg anatoli Belarus gmi 2393 +1 8 -3 valneris guntis Latvia gmi 2388 -9 9 +2 prosman erno Netherlands gmi 2378 +2 10 +2 dolfing martin Netherlands gmi 2377 +3 11 +3 shaibakov ainur Russia gmi 2373 +6 12 -2 virny vadim Germany gmi 2370 -7 13 +3 amrillaev murodullo Russia gmi 2367 +3 14 -5 meurs pim Netherlands gmi 2364 -16 15 = clerc rob Netherlands gmi 2363 -3 16 -3 ivanov artem Ukraine mi 2361 -11 17 +2 cordier arnaud France gmi 2358 +7 18 -1 ndjofang jean marc Cameroon gmi 2357 +4 19 +3 heusdens ron Netherlands gmi 2351 +3 20 +4 jansen gerard Netherlands gmi 2348 +3 21 +7 kalmakov andrej Russia gmi 2346 +5 22 +1 vermin hans Switzerland gmi 2346 = 23 -2 n'diaye macodou Senegal gmi 2344 -5 24 +5 anikeev yuriy Ukraine gmi 2343 +5 25 -7 krajenbrink johan Netherlands gmi 2341 -11 26 +1 van den akker jeroen Netherlands gmi 2339 -3 27 +3 leesmann kaido Estonia gmi 2337 = 28 +3 misans roberts Latvia mi 2336 = 29 +3 kolesov gawril Russia mi 2334 = 30 -4 vatutin evgeni -

The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z
REVIEW pubs.acs.org/CR The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease Jonathan Z. Long* and Benjamin F. Cravatt* The Skaggs Institute for Chemical Biology and Department of Chemical Physiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, United States CONTENTS 2.4. Other Phospholipases 6034 1. Introduction 6023 2.4.1. LIPG (Endothelial Lipase) 6034 2. Small-Molecule Hydrolases 6023 2.4.2. PLA1A (Phosphatidylserine-Specific 2.1. Intracellular Neutral Lipases 6023 PLA1) 6035 2.1.1. LIPE (Hormone-Sensitive Lipase) 6024 2.4.3. LIPH and LIPI (Phosphatidic Acid-Specific 2.1.2. PNPLA2 (Adipose Triglyceride Lipase) 6024 PLA1R and β) 6035 2.1.3. MGLL (Monoacylglycerol Lipase) 6025 2.4.4. PLB1 (Phospholipase B) 6035 2.1.4. DAGLA and DAGLB (Diacylglycerol Lipase 2.4.5. DDHD1 and DDHD2 (DDHD Domain R and β) 6026 Containing 1 and 2) 6035 2.1.5. CES3 (Carboxylesterase 3) 6026 2.4.6. ABHD4 (Alpha/Beta Hydrolase Domain 2.1.6. AADACL1 (Arylacetamide Deacetylase-like 1) 6026 Containing 4) 6036 2.1.7. ABHD6 (Alpha/Beta Hydrolase Domain 2.5. Small-Molecule Amidases 6036 Containing 6) 6027 2.5.1. FAAH and FAAH2 (Fatty Acid Amide 2.1.8. ABHD12 (Alpha/Beta Hydrolase Domain Hydrolase and FAAH2) 6036 Containing 12) 6027 2.5.2. AFMID (Arylformamidase) 6037 2.2. Extracellular Neutral Lipases 6027 2.6. Acyl-CoA Hydrolases 6037 2.2.1. PNLIP (Pancreatic Lipase) 6028 2.6.1. FASN (Fatty Acid Synthase) 6037 2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic 2.6.2. -

Lisa Strother Phd Thesis
INVESTIGATION OF MOLECULAR AND CELLULAR MECHANISMS UNDERPINNING THE NEUROTOXICITY OF HOMOCYSTEINE AND ITS METABOLITES IN MODELS OF NEURODEGENERATION Lisa Strother A Thesis Submitted for the Degree of PhD at the University of St Andrews 2018 Full metadata for this item is available in St Andrews Research Repository at: http://research-repository.st-andrews.ac.uk/ Please use this identifier to cite or link to this item: http://hdl.handle.net/10023/17067 This item is protected by original copyright Investigation of molecular and cellular mechanisms underpinning the neurotoxicity of homocysteine and its metabolites in models of neurodegeneration Lisa Strother CONTENTS LIST OF FIGURES ................................................................................................................. vii ACKNOWLEDGEMENTS ....................................................................................................... x ABSTRACT ............................................................................................................................. xii DECLARATIONS .................................................................................................................. xiv LIST OF ABBREVIATIONS ................................................................................................ xvii CHAPTER 1: INTRODUCTION AND BACKGROUND ....................................................... 1 1.1 INTRODUCTION ............................................................................................................ 2 1.1.1 Homocysteine -
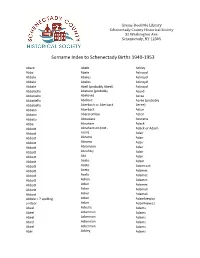
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -

Protein Network Analyses of Pulmonary Endothelial Cells In
www.nature.com/scientificreports OPEN Protein network analyses of pulmonary endothelial cells in chronic thromboembolic pulmonary hypertension Sarath Babu Nukala1,8,9*, Olga Tura‑Ceide3,4,5,9, Giancarlo Aldini1, Valérie F. E. D. Smolders2,3, Isabel Blanco3,4, Victor I. Peinado3,4, Manuel Castell6, Joan Albert Barber3,4, Alessandra Altomare1, Giovanna Baron1, Marina Carini1, Marta Cascante2,7,9 & Alfonsina D’Amato1,9* Chronic thromboembolic pulmonary hypertension (CTEPH) is a vascular disease characterized by the presence of organized thromboembolic material in pulmonary arteries leading to increased vascular resistance, heart failure and death. Dysfunction of endothelial cells is involved in CTEPH. The present study describes for the frst time the molecular processes underlying endothelial dysfunction in the development of the CTEPH. The advanced analytical approach and the protein network analyses of patient derived CTEPH endothelial cells allowed the quantitation of 3258 proteins. The 673 diferentially regulated proteins were associated with functional and disease protein network modules. The protein network analyses resulted in the characterization of dysregulated pathways associated with endothelial dysfunction, such as mitochondrial dysfunction, oxidative phosphorylation, sirtuin signaling, infammatory response, oxidative stress and fatty acid metabolism related pathways. In addition, the quantifcation of advanced oxidation protein products, total protein carbonyl content, and intracellular reactive oxygen species resulted increased -

Testicular Diffuse Large B-Cell Lymphoma—Clinical, Molecular, and Immunological Features
cancers Review Testicular Diffuse Large B-Cell Lymphoma—Clinical, Molecular, and Immunological Features Marjukka Pollari 1,2,* , Suvi-Katri Leivonen 1,3 and Sirpa Leppä 1,3 1 Research Program Unit, Faculty of Medicine, University of Helsinki, 00014 Helsinki, Finland; suvi-katri.leivonen@helsinki.fi (S.-K.L.); sirpa.leppa@helsinki.fi (S.L.) 2 Department of Oncology, Tays Cancer Center, Tampere University Hospital, 33521 Tampere, Finland 3 Department of Oncology, Comprehensive Cancer Center, Helsinki University Hospital, 00029 Helsinki, Finland * Correspondence: marjukka.pollari@pshp.fi Simple Summary: Testicular diffuse large B-cell lymphoma (T-DLBCL) is a rare and aggressive lymphoma entity that mainly affects elderly men. It has a high relapse rate with especially the relapses of the central nervous system associating with dismal outcome. T-DLBCL has a unique biology with distinct genetic characteristics and clinical presentation, and the increasing knowledge on the tumor microenvironment of T-DLBCL highlights the significance of the host immunity and immune escape in this rare lymphoma, presenting in an immune-privileged site of the testis. This review provides an update on the latest progress made in T-DLBCL research and summarizes the clinical perspectives in T-DLBCL. Abstract: Primary testicular lymphoma is a rare lymphoma entity, yet it is the most common testicular malignancy among elderly men. The majority of the cases represent non-germinal center B-cell- Citation: Pollari, M.; Leivonen, S.-K.; like (non-GCB) diffuse large B-cell lymphoma (DLBCL) with aggressive clinical behavior and a Leppä, S. Testicular Diffuse Large relatively high relapse rate. Due to the rareness of the disease, no randomized clinical trials have been B-Cell Lymphoma—Clinical, conducted and the currently recognized standard of care is based on retrospective analyses and few Molecular, and Immunological phase II trials. -

Epistatic Interactions Associated with Fatty Acid
FACULDADE DE CIÊNCIAS AGRÁRIAS E VETERINÁRIAS UNIVERSIDADE ESTADUAL PAULISTA CÂMPUS DE JABOTICABAL EPISTATIC INTERACTIONS ASSOCIATED WITH FATTY ACID PROFILE OF BEEF FROM NELLORE CATTLE Sabrina Thaise Amorim Zootecnista 2020 FACULDADE DE CIÊNCIAS AGRÁRIAS E VETERINÁRIAS UNIVERSIDADE ESTADUAL PAULISTA CÂMPUS DE JABOTICABAL EPISTATIC INTERACTIONS ASSOCIATED WITH FATTY ACID PROFILE OF BEEF FROM NELLORE CATTLE Sabrina Thaise Amorim Orientador: Prof. Dr. Fernando Sebastián Baldi Rey Co-orientador: Dr. Fernando Brito Lopes Co-orientadora: Dra. Nedenia Bonvino Stafuzza Dissertação apresentada à Faculdade de Ciências Agrárias e Veterinárias – Unesp, Campus de Jaboticabal, como parte das exigências para a obtenção do título de Mestre em Genética e Melhoramento Animal. 2020 Amorim, Sabrina Thaise A524e Epistatic interactions associated with fatty acid profile of beef from Nellore cattle / Sabrina Thaise Amorim. -- Jaboticabal, 2020 173 p. Dissertação (mestrado) - Universidade Estadual Paulista (Unesp), Faculdade de Ciências Agrárias e Veterinárias, Jaboticabal Orientador: Fernando Sebastián Baldi Rey Coorientador: Fernando Brito Lopes 1. Genética. 2. Melhoramento Animal. 3. Genômica. I. Título. Sistema de geração automática de fichas catalográficas da Unesp. Biblioteca da Faculdade de Ciências Agrárias e Veterinárias, Jaboticabal. Dados fornecidos pelo autor(a). Essa ficha não pode ser modificada. DADOS CURRICULARES DO AUTOR Sabrina Thaise Amorim, nascida em 02 de julho de 1995 na cidade de Brusque – Santa Catarina, filha de Alexandre Adriano Amorim e Liliane RaQuel Pavesi Amorim. Iniciou em março de 2013 o curso de graduação em Zootecnia na Universidade Federal de Santa Catarina, obtendo o título de Zootecnista em dezembro de 2017. Durante a graduação foi bolsista de Iniciação Científica do CNPq, monitora da disciplina “Genética Aplicada à Zootecnia”, integrante do Grupo de Pesquisa em Produção Animal e integrante do Laboratório de Pesquisa e Ensino em Genética Animal (LEPGA) da mesma instituição de fomento, sob a orientação do Prof. -

Acta Okl1-2014.Cdr
ACTA POLONIAE PHARMACEUTICA VOL. 72 No. 1 January/February 2015 ISSN 2353-5288 Drug Research EDITOR Aleksander P. Mazurek National Medicines Institute, The Medical University of Warsaw ASSISTANT EDITOR Jacek Bojarski Medical College, Jagiellonian University, KrakÛw EXECUTIVE EDITORIAL BOARD Miros≥awa Furmanowa The Medical University of Warsaw Boøenna Gutkowska The Medical University of Warsaw Roman Kaliszan The Medical University of GdaÒsk Jan Pachecka The Medical University of Warsaw Jan Pawlaczyk K. Marcinkowski University of Medical Sciences, PoznaÒ Janusz Pluta The Medical University of Wroc≥aw Witold Wieniawski Polish Pharmaceutical Society, Warsaw Pavel Komarek Czech Pharmaceutical Society Henry Ostrowski-Meissner Charles Sturt University, Sydney Erhard Rˆder Pharmazeutisches Institut der Universit‰t, Bonn Phil Skolnick DOV Pharmaceutical, Inc. Zolt·n Vincze Semmelweis University of Medicine, Budapest This Journal is published bimonthly by the Polish Pharmaceutical Society (Issued since 1937) The paper version of the Publisher magazine is a prime version. The electronic version can be found in the Internet on page www.actapoloniaepharmaceutica.pl An access to the journal in its electronics version is free of charge Impact factor (2013): 0.693 MNiSW score (2013): 15 points Index Copernicus (2012): 13.18 Charges Annual subscription rate for 2014 is US $ 210 including postage and handling charges. Prices subject to change. Back issues of previously published volumes are available directly from Polish Pharmaceutical Society, 16 D≥uga St., 00-238 Warsaw, Poland. Payment should be made either by bankerís draft (money order) issued to ÑPTFarmî or to our account Millennium S.A. No. 29 1160 2202 0000 0000 2770 0281, Polskie Towarzystwo Farmaceutyczne, ul. -

Thèse De Doctorat
UNIVERSITÉ PARIS-SUD ÉCOLE DOCTORALE 418 : DE CANCÉROLOGIE Laboratoire : Oncogenèse des épithéliums digestifs – Institut Cochin THÈSE DE DOCTORAT SCIENCES DE LA VIE ET DE LA SANTÉ par Pierre-Alexandre JUST Etude du rôle de LKB1 dans le foie Date de soutenance : 10/12/2014 Composition du jury : Directeur de thèse : Christine PERRET DR1, INSERM, Paris Rapporteurs : Valérie PARADIS PU-PH, Université Paris Diderot, Paris Anne-Françoise BURNOL DR1, CNRS, Paris Examinateurs : Benoît TERRIS PU-PH, Université Paris Descartes, Paris Jean ROSENBAUM DR1, INSERM, Bordeaux Président : Christian AUCLAIR PU, ENS, Cachan ii Remerciements Au moment d’achever la rédaction de ce mémoire, je souhaite remercier un grand nombre de personnes qui ont permis cette aventure dans le monde LKB1. Monsieur le professeur Christian Auclair, depuis mon Master 2, je suis étu- diant au sein de votre École doctorale et j’ai pu apprécier l’incroyable diversité, la pluridisciplinarité et la qualité de son enseignement. Vous avez à plusieurs reprises jugé mon travail lors de différentes présentations et vos remarques m’ont toujours été sources de réflexions fructueuses. Soyez-en remercié. Madame le professeur Valérie Paradis, vous me faites l’honneur de juger mon travail. Votre travail de recherche est en effet pour moi un exemple, alliant intelligemment observations anatomocliniques et confrontations biologiques. J’ap- précie votre raisonnement, partant d’approches morphologiques et aboutissant à de nouveaux paradigmes pour la carcinogenèse hépatique, notamment celle liée au syndrome métabolique. Soyez-en remerciée. Chère Anne-Françoise, voilà quelques années que je fréquente épisodique- ment ton bureau pour bénéficier de tes discussions pertinentes. Tu t’es toujours rendue disponible pour entendre mes hypothèses, souvent farfelues, et apporter, sans aucun a priori (ce qui est une qualité malheureusement trop rare dans le monde de la recherche), des éléments de réponse et des conseils expérimentaux de premier choix.