Lisa Strother Phd Thesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aspartic Acid Agonist, in the Mammalian Striatum
The Journal of Neuroscience August 1986, 6(8): 2226-2234 /II Vitro Release and Electrophysiological Effects In Situ of Homocysteic Acid, An Endogenous N-Methyl-(D)-aspartic Acid Agonist, in the Mammalian Striatum Kim Quang DO,* Paul L. Herrling,? Peter Streit,* Waldemar A. Turski,“fsl and Michel Cuenod* *Brain Research Institute, University of Zurich, Zurich, Switzerland, and j-Wander Research Institute, Bern, Switzerland A potassium-induced, calcium-dependent release of endogenous fects of microiontophoretically applied (L)-HCA on membrane homocysteic acid (HCA) from rat striatal slices was demonstrat- potential and cortically evoked EPSPsin cat caudate neurons, ed. A precolumn derivatization high-performance liquid chro- and the pharmacologicalspecificity of (L)-HCA in this structure. matography method was developed that allowed quantitative de- termination of sulfur-containing amino acids at the picomole level. Materials and Methods Intracellular recordings from cat caudate neurons during si- Materials multaneous microiontophoretic application of drugs and electri- cal stimulation of the corticocaudate pathway showed that (L> Release HCA evoked a depolarization pattern similar to that induced 4-N,N-Dimethylamino-azobenzene-4’-isotbiocyanate (DABITC) was ob- by %methyl-(D>aspartic acid (NMDA), and both these depo- tained from Fluka (Buchs, CH) and recrystallized in acetone (Merck, larizations could be selectively inhibited by a specific NMDA Darmstadt, FRG). All other solvents used were ofcommercial analytical antagonist, (D)-Z-amino-7-phosphonoheptanoicacid [(D)-AP-~]. grade from Merck or Fluka. The internal standard (D,L)-2-amino-7- A selective antagonismof (rJ-HCA-induced depolarizations by sulfonoheptanoic acid (AS-7) was a generous gift of Dr. J. C. Watkins. (L)-homocysteic acid [(L)-HCA], (L)-cysteine sulfinic acid [(L)-CSA], (L)- (D>AP-~ was confirmed in quantitative experiments with the cysteic acid [(L)-CA], and veratrine were purchased from Sigma (St. -

Treatment Protocol Copyright © 2018 Kostoff Et Al
Prevention and reversal of Alzheimer's disease: treatment protocol Copyright © 2018 Kostoff et al PREVENTION AND REVERSAL OF ALZHEIMER'S DISEASE: TREATMENT PROTOCOL by Ronald N. Kostoffa, Alan L. Porterb, Henry. A. Buchtelc (a) Research Affiliate, School of Public Policy, Georgia Institute of Technology, USA (b) Professor Emeritus, School of Public Policy, Georgia Institute of Technology, USA (c) Associate Professor, Department of Psychiatry, University of Michigan, USA KEYWORDS Alzheimer's Disease; Dementia; Text Mining; Literature-Based Discovery; Information Technology; Treatments Prevention and reversal of Alzheimer's disease: treatment protocol Copyright © 2018 Kostoff et al CITATION TO MONOGRAPH Kostoff RN, Porter AL, Buchtel HA. Prevention and reversal of Alzheimer's disease: treatment protocol. Georgia Institute of Technology. 2018. PDF. https://smartech.gatech.edu/handle/1853/59311 COPYRIGHT AND CREATIVE COMMONS LICENSE COPYRIGHT Copyright © 2018 by Ronald N. Kostoff, Alan L. Porter, Henry A. Buchtel Printed in the United States of America; First Printing, 2018 CREATIVE COMMONS LICENSE This work can be copied and redistributed in any medium or format provided that credit is given to the original author. For more details on the CC BY license, see: http://creativecommons.org/licenses/by/4.0/ This work is licensed under a Creative Commons Attribution 4.0 International License<http://creativecommons.org/licenses/by/4.0/>. DISCLAIMERS The views in this monograph are solely those of the authors, and do not represent the views of the Georgia Institute of Technology or the University of Michigan. This monograph is not intended as a substitute for the medical advice of physicians. The reader should regularly consult a physician in matters relating to his/her health and particularly with respect to any symptoms that may require diagnosis or medical attention. -

Endogenous Metabolites: JHU NIMH Center Page 1
S. No. Amino Acids (AA) 24 L-Homocysteic acid 1 Glutaric acid 25 L-Kynurenine 2 Glycine 26 N-Acetyl-Aspartic acid 3 L-arginine 27 N-Acetyl-L-alanine 4 L-Aspartic acid 28 N-Acetyl-L-phenylalanine 5 L-Glutamine 29 N-Acetylneuraminic acid 6 L-Histidine 30 N-Methyl-L-lysine 7 L-Isoleucine 31 N-Methyl-L-proline 8 L-Leucine 32 NN-Dimethyl Arginine 9 L-Lysine 33 Norepinephrine 10 L-Methionine 34 Phenylacetyl-L-glutamine 11 L-Phenylalanine 35 Pyroglutamic acid 12 L-Proline 36 Sarcosine 13 L-Serine 37 Serotonin 14 L-Tryptophan 38 Stachydrine 15 L-Tyrosine 39 Taurine 40 Urea S. No. AA Metabolites and Conjugates 1 1-Methyl-L-histidine S. No. Carnitine conjugates 2 2-Methyl-N-(4-Methylphenyl)alanine 1 Acetyl-L-carnitine 3 3-Methylindole 2 Butyrylcarnitine 4 3-Methyl-L-histidine 3 Decanoyl-L-carnitine 5 4-Aminohippuric acid 4 Isovalerylcarnitine 6 5-Hydroxylysine 5 Lauroyl-L-carnitine 7 5-Hydroxymethyluracil 6 L-Glutarylcarnitine 8 Alpha-Aspartyl-lysine 7 Linoleoylcarnitine 9 Argininosuccinic acid 8 L-Propionylcarnitine 10 Betaine 9 Myristoyl-L-carnitine 11 Betonicine 10 Octanoylcarnitine 12 Carnitine 11 Oleoyl-L-carnitine 13 Creatine 12 Palmitoyl-L-carnitine 14 Creatinine 13 Stearoyl-L-carnitine 15 Dimethylglycine 16 Dopamine S. No. Krebs Cycle 17 Epinephrine 1 Aconitate 18 Hippuric acid 2 Citrate 19 Homo-L-arginine 3 Ketoglutarate 20 Hydroxykynurenine 4 Malate 21 Indolelactic acid 5 Oxalo acetate 22 L-Alloisoleucine 6 Succinate 23 L-Citrulline 24 L-Cysteine-glutathione disulfide Semi-quantitative analysis of endogenous metabolites: JHU NIMH Center Page 1 25 L-Glutathione, reduced Table 1: Semi-quantitative analysis of endogenous molecules and their derivatives by Liquid Chromatography- Mass Spectrometry (LC-TripleTOF “or” LC-QTRAP). -

Involvements of Hyperhomocysteinemia in Neurological Disorders
H OH metabolites OH Review Involvements of Hyperhomocysteinemia in Neurological Disorders Marika Cordaro 1,† , Rosalba Siracusa 2,† , Roberta Fusco 2 , Salvatore Cuzzocrea 2,3,* , Rosanna Di Paola 2,* and Daniela Impellizzeri 2 1 Department of Biomedical, Dental and Morphological and Functional Imaging, University of Messina, Via Consolare Valeria, 98125 Messina, Italy; [email protected] 2 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, 98166 Messina, Italy; [email protected] (R.S.); [email protected] (R.F.); [email protected] (D.I.) 3 Department of Pharmacological and Physiological Science, Saint Louis University School of Medicine, Saint Louis, MO 63104, USA * Correspondence: [email protected] (S.C.); [email protected] (R.D.P.); Tel.: +39-090-6765208 (S.C. & R.D.P.) † The authors equally contributed to the review. Abstract: Homocysteine (HCY), a physiological amino acid formed when proteins break down, leads to a pathological condition called hyperhomocysteinemia (HHCY), when it is over a definite limit. It is well known that an increase in HCY levels in blood, can contribute to arterial damage and several cardiovascular disease, but the knowledge about the relationship between HCY and brain disorders is very poor. Recent studies demonstrated that an alteration in HCY metabolism or a deficiency in folate or vitamin B12 can cause altered methylation and/or redox potentials, that leads to a modification on calcium influx in cells, or into an accumulation in amyloid and/or tau protein involving a cascade of events that culminate in apoptosis, and, in the worst conditions, neuronal death. The present review will thus summarize how much is known about the possible role of HHCY in neurodegenerative disease. -

Primordial Synthesis of Amines and Amino Acids in a 1958 Miller H2 S
Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment Eric T. Parkera,1, Henderson J. Cleavesb, Jason P. Dworkinc, Daniel P. Glavinc, Michael Callahanc, Andrew Aubreyd, Antonio Lazcanoe, and Jeffrey L. Badaa,2 aGeosciences Research Division, Scripps Institution of Oceanography, University of California at San Diego, 8615 Kennel Way, La Jolla, CA 92093; bGeophysical Laboratory, Carnegie Institution of Washington, 5251 Broad Branch Road NW, Washington, DC 20015; cNational Aeronautics and Space Administration Goddard Space Flight Center, Solar System Exploration Division, Greenbelt, MD 20771; dNational Aeronautics and Space Administration Jet Propulsion Laboratory, California Institute of Technology, 4800 Oak Grove Drive, Pasadena, CA 91109; and eFacultad de Ciencias, Universidad Nacional Autónoma de México, Apdo. Postal 70-407 Ciudad Universitaria, 04510 Mexico D.F., Mexico Edited by Gerald F. Joyce, The Scripps Research Institute, La Jolla, CA, and approved February 14, 2011 (received for review December 22, 2010) Archived samples from a previously unreported 1958 Stanley Miller known volcanic apparatus were found to contain a wide variety of electric discharge experiment containing hydrogen sulfide (H2S) amino acids and amines, including ornithine, homoserine, methy- were recently discovered and analyzed using high-performance lamine, and ethylamine, many of which had not been reported liquid chromatography and time-of-flight mass spectrometry. We previously in spark discharge experiments (7). The volcanic report here the detection and quantification of primary amine- apparatus differed from Miller’s classic apparatus in that it uti- containing compounds in the original sample residues, which were lized an aspirator that injected steam into the electric discharge produced via spark discharge using a gaseous mixture of H2S, CH4, chamber, simulating a volcanic eruption (1). -

The Controversial Role of Homocysteine in Neurology: from Labs to Clinical Practice
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archivio istituzionale della ricerca - Università di Trieste International Journal of Molecular Sciences Review The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice Rita Moretti * and Paola Caruso Neurology Clinic, Department of Medical, Surgical, and Health Sciences, University of Trieste, 34149 Trieste, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-040-399-4572 Received: 29 November 2018; Accepted: 4 January 2019; Published: 8 January 2019 Abstract: Homocysteine (Hcy) is a sulfur-containing amino acid that is generated during methionine metabolism. Physiologic Hcy levels are determined primarily by dietary intake and vitamin status. Elevated plasma levels of Hcy can be caused by deficiency of either vitamin B12 or folate. Hyperhomocysteinemia (HHcy) can be responsible of different systemic and neurological disease. Actually, HHcy has been considered as a risk factor for systemic atherosclerosis and cardiovascular disease (CVD) and HHcy has been reported in many neurologic disorders including cognitive impairment and stroke, independent of long-recognized factors such as hyperlipidemia, hypertension, diabetes mellitus, and smoking. HHcy is typically defined as levels >15 micromol/L. Treatment of hyperhomocysteinemia with folic acid and B vitamins seems to be effective in the prevention of the development of atherosclerosis, CVD, and strokes. However, data from literature show controversial results regarding the significance of homocysteine as a risk factor for CVD and stroke and whether patients should be routinely screened for homocysteine. HHcy-induced oxidative stress, endothelial dysfunction, inflammation, smooth muscle cell proliferation, and endoplasmic reticulum (ER) stress have been considered to play an important role in the pathogenesis of several diseases including atherosclerosis and stroke. -

2013 Kaiser Permanente-Authored Publications Alphabetical by Author
2013 Kaiser Permanente-Authored Publications Alphabetical by Author 1. Abbas MA, Cannom RR, Chiu VY, Burchette RJ, Radner GW, Haigh PI, Etzioni DA Triage of patients with acute diverticulitis: are some inpatients candidates for outpatient treatment? Colorectal Dis. 2013 Apr;15(4):451-7. Southern California 23061533 KP author(s): Abbas, Maher A; Chiu, Vicki Y; Burchette, Raoul J; Radner, Gary W; Haigh, Philip I 2. Abbass MA, Slezak JM, DiFronzo LA Predictors of Early Postoperative Outcomes in 375 Consecutive Hepatectomies: A Single- institution Experience Am Surg. 2013 Oct;79(10):961-7. Southern California 24160779 KP author(s): Abbass, Mohammad Ali; Slezak, Jeffrey M; DiFronzo, Andrew L 3. Abbenhardt C, Poole EM, Kulmacz RJ, Xiao L, Curtin K, Galbraith RL, Duggan D, Hsu L, Makar KW, Caan BJ, Koepl L, Owen RW, Scherer D, Carlson CS, Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) and CCFR, Potter JD, Slattery ML, Ulrich CM Phospholipase A2G1B polymorphisms and risk of colorectal neoplasia Int J Mol Epidemiol Genet. 2013 Sep 12;4(3):140-9. Northern California 24046806 KP author(s): Caan, Bette 4. Aberg JA, Gallant JE, Ghanem KG, Emmanuel P, Zingman BS, Horberg MA Primary Care Guidelines for the Management of Persons Infected With HIV: 2013 Update by the HIV Medicine Association of the Infectious Diseases Society of America Clin Infect Dis. 2013 Nov 13. Mid-Atlantic 24235263 KP author(s): Horberg, Michael 5. Abraham AG, D'Souza G, Jing Y, Gange SJ, Sterling TR, Silverberg MJ, Saag MS, Rourke SB, Rachlis A, Napravnik S, Moore RD, Klein MB, Kitahata MM, Kirk GD, Hogg RS, Hessol NA, Goedert JJ, Gill MJ, Gebo KA, Eron JJ, Engels EA, Dubrow R, Crane HM, Brooks JT, Bosch RJ, Strickler HD, North American AIDS Cohort Collaboration on Research and Design of IeDEA Invasive cervical cancer risk among HIV-infected women: A North American multi-cohort collaboration prospective study J Acquir Immune Defic Syndr. -

World Ratings Draughts
FMJD Rating No 83 January/March 2015 FMJD rating Nr 83 January 2015 email: [email protected] 1 FMJD Rating No 83 January/March 2015 With any question concerning this issue You can contact me directly via email: [email protected] 2 FMJD Rating No 83 January/March 2015 1. Rating - open Name Nationality Title Rating +/- 1 = shvartsman alexander Russia gmi 2425 -19 2 = georgiev alexander Russia gmi 2421 -15 3 +5 chizhov alexey Russia gmi 2409 +18 4 = getmanski alexander Russia gmi 2404 +4 5 +1 boomstra roel Netherlands gmi 2399 +4 6 -3 baliakin alexander Netherlands gmi 2396 -11 7 = gantvarg anatoli Belarus gmi 2393 +1 8 -3 valneris guntis Latvia gmi 2388 -9 9 +2 prosman erno Netherlands gmi 2378 +2 10 +2 dolfing martin Netherlands gmi 2377 +3 11 +3 shaibakov ainur Russia gmi 2373 +6 12 -2 virny vadim Germany gmi 2370 -7 13 +3 amrillaev murodullo Russia gmi 2367 +3 14 -5 meurs pim Netherlands gmi 2364 -16 15 = clerc rob Netherlands gmi 2363 -3 16 -3 ivanov artem Ukraine mi 2361 -11 17 +2 cordier arnaud France gmi 2358 +7 18 -1 ndjofang jean marc Cameroon gmi 2357 +4 19 +3 heusdens ron Netherlands gmi 2351 +3 20 +4 jansen gerard Netherlands gmi 2348 +3 21 +7 kalmakov andrej Russia gmi 2346 +5 22 +1 vermin hans Switzerland gmi 2346 = 23 -2 n'diaye macodou Senegal gmi 2344 -5 24 +5 anikeev yuriy Ukraine gmi 2343 +5 25 -7 krajenbrink johan Netherlands gmi 2341 -11 26 +1 van den akker jeroen Netherlands gmi 2339 -3 27 +3 leesmann kaido Estonia gmi 2337 = 28 +3 misans roberts Latvia mi 2336 = 29 +3 kolesov gawril Russia mi 2334 = 30 -4 vatutin evgeni -

The Overlooked Modulating Role of Nitrous Oxide for Corrinoid-Dependent Microbial Processes
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2019 THE OVERLOOKED MODULATING ROLE OF NITROUS OXIDE FOR CORRINOID-DEPENDENT MICROBIAL PROCESSES Yongchao Yin University of Tennessee, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Recommended Citation Yin, Yongchao, "THE OVERLOOKED MODULATING ROLE OF NITROUS OXIDE FOR CORRINOID- DEPENDENT MICROBIAL PROCESSES. " PhD diss., University of Tennessee, 2019. https://trace.tennessee.edu/utk_graddiss/5761 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yongchao Yin entitled "THE OVERLOOKED MODULATING ROLE OF NITROUS OXIDE FOR CORRINOID-DEPENDENT MICROBIAL PROCESSES." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Microbiology. Frank Löffler, Major Professor We have read this dissertation and recommend its acceptance: Gary Sayler, Alison Buchan, Qiang He Accepted for the Council: Dixie L. Thompson Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) THE OVERLOOKED MODULATING ROLE OF NITROUS OXIDE FOR CORRINOID-DEPENDENT MICROBIAL PROCESSES A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Yongchao Yin December 2019 Copyright © 2019 by Yongchao Yin. -

Ageing Research Reviews 49 (2019) 144–164
Ageing Research Reviews 49 (2019) 144–164 Contents lists available at ScienceDirect Ageing Research Reviews journal homepage: www.elsevier.com/locate/arr Homocysteine and age-associated disorders T ⁎ E.A. Ostrakhovitch , S. Tabibzadeh Frontiers in Bioscience Research Institute in Aging and Cancer, Irvine, CA, USA ARTICLE INFO ABSTRACT Keywords: There are numerous theories of aging, a process which still seems inevitable. Aging leads to cancer and multi- Homocysteine systemic disorders as well as chronic diseases. Decline in age- associated cellular functions leads to neurode- Hyperhomocysteinemia generation and cognitive decline that affect the quality of life. Accumulation of damage, mutations, metabolic Metabolism changes, failure in cellular energy production and clearance of altered proteins over the lifetime, and hy- Aging perhomocysteinemia, ultimately result in tissue degeneration. The decline in renal functions, nutritional defi- Age associated diseases ciencies, deregulation of methionine cycle and deficiencies of homocysteine remethylation and transsulfuration cofactors cause elevation of homocysteine with advancing age. Abnormal accumulation of homocysteine is a risk factor of cardiovascular, neurodegenerative and chronic kidney disease. Moreover, approximately 50% of people, aged 65 years and older develop hypertension and are at a high risk of developing cardiovascular in- sufficiency and incurable neurodegenerative disorders. Increasing evidence suggests inverse relation between cognitive impairment, cerebrovascular and cardiovascular events and renal function. Oxidative stress, in- activation of nitric oxide synthase pathway and mitochondria dysfunction associated with impaired homo- cysteine metabolism lead to aging tissue degeneration. In this review, we examine impact of high homocysteine levels on changes observed with aging that contribute to development and progression of age associated dis- eases. -
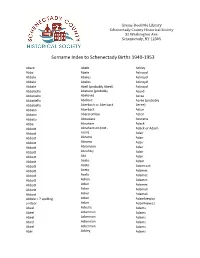
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -

Acetylcholine Release from the Rabbit Retina Mediated by Kainate Receptors
The Journal of Neuroscience, January 1991, 7 f(1): 11 l-l 22 Acetylcholine Release from the Rabbit Retina Mediated by Kainate Receptors David M. Linn,’ Christine Blazynski,* Dianna A. Redburn, and Stephen C. Massey’ Sensory Sciences Center, Graduate School of Biomedical Sciences, University of Texas Health Science Center, Houston, Texas 77030, 2Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, St. Louis, Missouri 63110, and 3Deoartment of Neurobiology and Anatomy, University of Texas Medical School, Houston,.Texas 77225 The cholinergic amacrine cells of the rabbit retina may be KA or QQ receptors (Slaughter and Miller, 1983a,b; Copen- labeled with 3H-choline (3H-Ch), and the activity of the cho- hagen and Jahr, 1989). In contrast, the sign-inverting input to linergic population may be monitored by following the re- ON bipolar cells is mediated by APB receptors (Shiells et al., lease of H-ACh. Glutamate analogs caused massive ACh 198 1; Slaughter and Miller, 1981, 1985). In fact, APB is a spe- release, up to 50 times the basal efflux, with the following cific agonist at this site, which underlies the separation of ON rank order of potency: a-amino-3-hydroxy-5-methyl-4-isox- and OFF responsesthroughout the visual system.Although KA, azolepropionic acid (AMPA) > quisqualate (QQ) = kainate QQ, and APB receptors have been found in the outer retina, (KA) z+ NMDA (in magnesium-free medium) B glutamate > NMDA receptors appear to be rare or absent. NMDA and aspartate. In contrast, the release of 3H-Ch was unchanged. NMDA antagonistshave little or no effect on second-orderneu- Submaximal doses of each agonist were used to establish rons of most species,including the rabbit (Shiells et al., 1981; the specifity of glutamate antagonists.