Supporting Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metaproteogenomic Insights Beyond Bacterial Response to Naphthalene
ORIGINAL ARTICLE ISME Journal – Original article Metaproteogenomic insights beyond bacterial response to 5 naphthalene exposure and bio-stimulation María-Eugenia Guazzaroni, Florian-Alexander Herbst, Iván Lores, Javier Tamames, Ana Isabel Peláez, Nieves López-Cortés, María Alcaide, Mercedes V. del Pozo, José María Vieites, Martin von Bergen, José Luis R. Gallego, Rafael Bargiela, Arantxa López-López, Dietmar H. Pieper, Ramón Rosselló-Móra, Jesús Sánchez, Jana Seifert and Manuel Ferrer 10 Supporting Online Material includes Text (Supporting Materials and Methods) Tables S1 to S9 Figures S1 to S7 1 SUPPORTING TEXT Supporting Materials and Methods Soil characterisation Soil pH was measured in a suspension of soil and water (1:2.5) with a glass electrode, and 5 electrical conductivity was measured in the same extract (diluted 1:5). Primary soil characteristics were determined using standard techniques, such as dichromate oxidation (organic matter content), the Kjeldahl method (nitrogen content), the Olsen method (phosphorus content) and a Bernard calcimeter (carbonate content). The Bouyoucos Densimetry method was used to establish textural data. Exchangeable cations (Ca, Mg, K and 10 Na) extracted with 1 M NH 4Cl and exchangeable aluminium extracted with 1 M KCl were determined using atomic absorption/emission spectrophotometry with an AA200 PerkinElmer analyser. The effective cation exchange capacity (ECEC) was calculated as the sum of the values of the last two measurements (sum of the exchangeable cations and the exchangeable Al). Analyses were performed immediately after sampling. 15 Hydrocarbon analysis Extraction (5 g of sample N and Nbs) was performed with dichloromethane:acetone (1:1) using a Soxtherm extraction apparatus (Gerhardt GmbH & Co. -

Characterization of the Interaction Between R. Conorii and Human
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 4-5-2018 Characterization of the Interaction Between R. Conorii and Human Host Vitronectin in Rickettsial Pathogenesis Abigail Inez Fish Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Bacteria Commons, Bacteriology Commons, Biology Commons, Immunology of Infectious Disease Commons, and the Pathogenic Microbiology Commons Recommended Citation Fish, Abigail Inez, "Characterization of the Interaction Between R. Conorii and Human Host Vitronectin in Rickettsial Pathogenesis" (2018). LSU Doctoral Dissertations. 4566. https://digitalcommons.lsu.edu/gradschool_dissertations/4566 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. CHARACTERIZATION OF THE INTERACTION BETWEEN R. CONORII AND HUMAN HOST VITRONECTIN IN RICKETTSIAL PATHOGENESIS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Interdepartmental Program in Biomedical and Veterinary Medical Sciences Through the Department of Pathobiological Sciences by Abigail Inez -

Evolutionary Origin of Insect–Wolbachia Nutritional Mutualism
Evolutionary origin of insect–Wolbachia nutritional mutualism Naruo Nikoha,1, Takahiro Hosokawab,1, Minoru Moriyamab,1, Kenshiro Oshimac, Masahira Hattoric, and Takema Fukatsub,2 aDepartment of Liberal Arts, The Open University of Japan, Chiba 261-8586, Japan; bBioproduction Research Institute, National Institute of Advanced Industrial Science and Technology, Tsukuba 305-8566, Japan; and cCenter for Omics and Bioinformatics, Graduate School of Frontier Sciences, University of Tokyo, Kashiwa 277-8561, Japan Edited by Nancy A. Moran, University of Texas at Austin, Austin, TX, and approved June 3, 2014 (received for review May 20, 2014) Obligate insect–bacterium nutritional mutualism is among the insects, generally conferring negative fitness consequences to most sophisticated forms of symbiosis, wherein the host and the their hosts and often causing hosts’ reproductive aberrations to symbiont are integrated into a coherent biological entity and un- enhance their own transmission in a selfish manner (7, 8). Re- able to survive without the partnership. Originally, however, such cently, however, a Wolbachia strain associated with the bedbug obligate symbiotic bacteria must have been derived from free-living Cimex lectularius,designatedaswCle, was shown to be es- bacteria. How highly specialized obligate mutualisms have arisen sential for normal growth and reproduction of the blood- from less specialized associations is of interest. Here we address this sucking insect host via provisioning of B vitamins (9). Hence, it –Wolbachia evolutionary -

Sanguineus Bacterial Communities Rhipicephalus
Composition and Seasonal Variation of Rhipicephalus turanicus and Rhipicephalus sanguineus Bacterial Communities Itai Lalzar, Shimon Harrus, Kosta Y. Mumcuoglu and Yuval Gottlieb Appl. Environ. Microbiol. 2012, 78(12):4110. DOI: 10.1128/AEM.00323-12. Published Ahead of Print 30 March 2012. Downloaded from Updated information and services can be found at: http://aem.asm.org/content/78/12/4110 These include: SUPPLEMENTAL MATERIAL Supplemental material http://aem.asm.org/ REFERENCES This article cites 44 articles, 17 of which can be accessed free at: http://aem.asm.org/content/78/12/4110#ref-list-1 CONTENT ALERTS Receive: RSS Feeds, eTOCs, free email alerts (when new articles cite this article), more» on June 10, 2013 by guest Information about commercial reprint orders: http://journals.asm.org/site/misc/reprints.xhtml To subscribe to to another ASM Journal go to: http://journals.asm.org/site/subscriptions/ Composition and Seasonal Variation of Rhipicephalus turanicus and Rhipicephalus sanguineus Bacterial Communities Itai Lalzar,a Shimon Harrus,a Kosta Y. Mumcuoglu,b and Yuval Gottlieba Koret School of Veterinary Medicine, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel,a and Department of Microbiology and Molecular Genetics, The Kuvin Center for the Study of Infectious and Tropical Diseases, Hadassah Medical School, The Institute for Medical Research Israel-Canada, The Hebrew University of Jerusalem, Jerusalem, Israelb A 16S rRNA gene approach, including 454 pyrosequencing and quantitative PCR (qPCR), was used to describe the bacterial com- munity in Rhipicephalus turanicus and to evaluate the dynamics of key bacterial tenants of adult ticks during the active questing season. -

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

Genome Project Reveals a Putative Rickettsial Endosymbiont
GBE Bacterial DNA Sifted from the Trichoplax adhaerens (Animalia: Placozoa) Genome Project Reveals a Putative Rickettsial Endosymbiont Timothy Driscoll1,y, Joseph J. Gillespie1,2,*,y, Eric K. Nordberg1,AbduF.Azad2, and Bruno W. Sobral1,3 1Virginia Bioinformatics Institute at Virginia Polytechnic Institute and State University 2Department of Microbiology and Immunology, University of Maryland School of Medicine 3Present address: Nestle´ Institute of Health Sciences SA, Campus EPFL, Quartier de L’innovation, Lausanne, Switzerland *Corresponding author: E-mail: [email protected]. yThese authors contributed equally to this work. Accepted: March 1, 2013 Abstract Eukaryotic genome sequencing projects often yield bacterial DNA sequences, data typically considered as microbial contamination. However, these sequences may also indicate either symbiont genes or lateral gene transfer (LGT) to host genomes. These bacterial sequences can provide clues about eukaryote–microbe interactions. Here, we used the genome of the primitive animal Trichoplax adhaerens (Metazoa: Placozoa), which is known to harbor an uncharacterized Gram-negative endosymbiont, to search for the presence of bacterial DNA sequences. Bioinformatic and phylogenomic analyses of extracted data from the genome assembly (181 bacterial coding sequences [CDS]) and trace read archive (16S rDNA) revealed a dominant proteobacterial profile strongly skewed to Rickettsiales (Alphaproteobacteria) genomes. By way of phylogenetic analysis of 16S rDNA and 113 proteins conserved across proteobacterial genomes, as well as identification of 27 rickettsial signature genes, we propose a Rickettsiales endosymbiont of T. adhaerens (RETA). The majority (93%) of the identified bacterial CDS belongs to small scaffolds containing prokaryotic-like genes; however, 12 CDS were identified on large scaffolds comprised of eukaryotic-like genes, suggesting that T. -

The Risk to Human Health from Free-Living Amoebae Interaction with Legionella in Drinking and Recycled Water Systems
THE RISK TO HUMAN HEALTH FROM FREE-LIVING AMOEBAE INTERACTION WITH LEGIONELLA IN DRINKING AND RECYCLED WATER SYSTEMS Dissertation submitted by JACQUELINE MARIE THOMAS BACHELOR OF SCIENCE (HONOURS) AND BACHELOR OF ARTS, UNSW In partial fulfillment of the requirements for the award of DOCTOR OF PHILOSOPHY in ENVIRONMENTAL ENGINEERING SCHOOL OF CIVIL AND ENVIRONMENTAL ENGINEERING FACULTY OF ENGINEERING MAY 2012 SUPERVISORS Professor Nicholas Ashbolt Office of Research and Development United States Environmental Protection Agency Cincinnati, Ohio USA and School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Professor Richard Stuetz School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Doctor Torsten Thomas School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales Sydney, Australia ORIGINALITY STATEMENT '1 hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom 1 have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.' Signed ~ ............................ -

18167638.Pdf
REVIEW ARTICLE Amoebal pathogens as emerging causal agents of pneumonia Fred´ eric´ Lamoth1 & Gilbert Greub1,2 1Infectious Diseases Service, University of Lausanne, Lausanne, Switzerland; and 2Center for Research on Intracellular Bacteria, Institute of Microbiology, University Hospital Center and University of Lausanne, Lausanne, Switzerland Correspondence: Gilbert Greub, Center for Abstract Research on Intracellular Bacteria, Institute of Microbiology, University Hospital Center and Despite using modern microbiological diagnostic approaches, the aetiological University of Lausanne, Rue du Bugnon 46, agents of pneumonia remain unidentified in about 50% of cases. Some bacteria 1011 Lausanne, Switzerland. Tel.: 141 21 that grow poorly or not at all in axenic media used in routine clinical bacteriology 31449 79; fax: 141 21 31440 60; e-mail: laboratory but which can develop inside amoebae may be the agents of these lower [email protected] respiratory tract infections (RTIs) of unexplained aetiology. Such amoebae- resisting bacteria, which coevolved with amoebae to resist their microbicidal Received 24 September 2009; revised 30 machinery, may have developed virulence traits that help them survive within November 2009; accepted 2 December 2009. human macrophages, i.e. the first line of innate immune defence in the lung. We Final version published online 22 January 2010. review here the current evidence for the emerging pathogenic role of various DOI:10.1111/j.1574-6976.2009.00207.x amoebae-resisting microorganisms as agents of RTIs in humans. Specifically, we discuss the emerging pathogenic roles of Legionella-like amoebal pathogens, novel Editor: Colin Berry Chlamydiae (Parachlamydia acanthamoebae, Simkania negevensis), waterborne mycobacteria and Bradyrhizobiaceae (Bosea and Afipia spp.). Keywords free-living amoebae; amoebae-resisting bacteria; Legionella; Chlamydia-like bacteria; mycobacteria; pneumonia. -

Paradoxical Evolution of Rickettsial Genomes
Ticks and Tick-borne Diseases 10 (2019) 462–469 Contents lists available at ScienceDirect Ticks and Tick-borne Diseases journal homepage: www.elsevier.com/locate/ttbdis Paradoxical evolution of rickettsial genomes T ⁎ Awa Diopa, Didier Raoultb, Pierre-Edouard Fourniera, a UMR VITROME, Aix-Marseille University, IRD, Service de Santé des Armées, Assistance Publique-Hôpitaux de Marseille, Institut Hospitalo-Uuniversitaire Méditerranée Infection, 19-21 Boulevard Jean Moulin, 13005, Marseille, France b UMR MEPHI, Aix-Marseille University, IRD, Assistance Publique-Hôpitaux de Marseille, Institut Hospitalo-Uuniversitaire Méditerranée Infection, Marseille, France ARTICLE INFO ABSTRACT Keywords: Rickettsia species are strictly intracellular bacteria that evolved approximately 150 million years ago from a Rickettsia presumably free-living common ancestor from the order Rickettsiales that followed a transition to an obligate Genomics intracellular lifestyle. Rickettsiae are best known as human pathogens vectored by various arthropods causing a Evolution range of mild to severe human diseases. As part of their obligate intracellular lifestyle, rickettsial genomes have Virulence undergone a convergent evolution that includes a strong genomic reduction resulting from progressive gene Genome rearrangement degradation, genomic rearrangements as well as a paradoxical expansion of various genetic elements, notably Non-coding DNA Gene loss small RNAs and short palindromic elements whose role remains unknown. This reductive evolutionary process is DNA repeats not unique to members of the Rickettsia genus but is common to several human pathogenic bacteria. Gene loss, gene duplication, DNA repeat duplication and horizontal gene transfer all have shaped rickettsial genome evolution. Gene loss mostly involved amino-acid, ATP, LPS and cell wall component biosynthesis and tran- scriptional regulators, but with a high preservation of toxin-antitoxin (TA) modules, recombination and DNA repair proteins. -

Detecting Phylogenetic Signals from Deep Roots of the Tree of Life
UNIVERSITY OF CALIFORNIA,MERCED Detecting Phylogenetic Signals From Deep Roots of the Tree of Life A dissertation submitted in partial fulfillment of the requirements for the degree Doctor of Philosophy in Quantitative and Systems Biology by Katherine Colleen Harris Amrine Committee in charge: Professor Carolin Frank, Chair Professor David Ardell Professor Meng-Lin Tsao Professor Suzanne Sindi August 2013 Copyright Katherine C. Amrine All Rights Reserved UNIVERSITY OF CALIFORNIA,MERCED Graduate Division The Dissertation of Katherine Colleen Harris Amrine is approved, and it is acceptable in quality and form for publication on microfilm and electronically: Faculty Advisor: David H. Ardell Committee Members: Chair: Carolin Frank Meng-Lin Tsao Suzanne Sindi Date iii Contents List of Figures ................................................................. vi List of Tables .................................................................. ix Acknowledgements ............................................................. x Vita ........................................................................... xi Abstract ...................................................................... xii 1 Shifting focus in evolutionary biology – identifying a new signal for phylogenetic tree reconstruction and taxonomic classification 1 1.1 The evolution of bacterial classification and phylogeny . .1 1.2 The historical marker – 16S . .2 1.3 Complications in bacterial classification and phylogeny . .2 1.3.1 Horizontal gene transfer . .2 1.3.2 Does a true tree exist? . .3 1.4 Methods for phylogenetic tree reconstruction . .3 1.4.1 DNA . .3 1.4.2 RNA . .4 1.4.3 Proteins . .4 1.4.4 Data compilation . .5 1.5 Bias in tree-building . .5 1.6 Biological bias in biological data . .6 1.7 The tRNA interaction network . .6 1.8 Information theory . .8 1.9 Machine Learning for bacterial classification . .9 2 tRNA signatures reveal polyphyletic origins of streamlined SAR11 genomes among the Alphaproteobacteria 12 2.1 Abstract . -
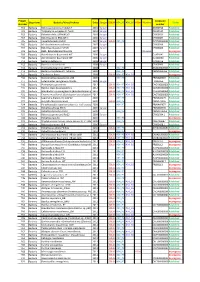
Project Number Organisms Bacteria/Virus/Archaea Date
Project_ Accession Organisms Bacteria/Virus/Archaea Date Sanger SOLiD 454_PE 454_SG PGM Illumina Status Number number P01 Bacteria Rickettsia conorii str.Malish 7 2001 Sanger AE006914 Published P02 Bacteria Tropheryma whipplei str.Twist 2003 Sanger AE014184 Published P03 Bacteria Rickettsia felis URRWXCal2 2005 Sanger CP000053 Published P04 Bacteria Rickettsia bellii RML369-C 2006 Sanger CP000087 Published P05 Bacteria Coxiella burnetii CB109 2007 Sanger SOLiD 454_PE AKYP00000000 Published P06 Bacteria Minibacterium massiliensis 2007 Sanger CP000269 Published P07 Bacteria Rickettsia massiliae MTU5 2007 Sanger CP000683 Published P08 Bacteria BaBL=Bête à Bernard Lascola 2007 Illumina In progress P09 Bacteria Acinetobacter baumannii AYE 2006 Sanger CU459141 Published P10 Bacteria Acinetobacter baumannii SDF 2006 Sanger CU468230 Published P11 Bacteria Borrelia duttonii Ly 2008 Sanger CP000976 Published P12 Bacteria Borrelia recurrentis A1 2008 Sanger CP000993 Published P13 Bacteria Francisella tularensis URFT1 2008 454_PE ABAZ00000000Published P14 Bacteria Borrelia crocidurae str. Achema 2009 454_PE PRJNA162335 Published P15 Bacteria Citrobacter koseri 2009 SOLiD 454_PE 454_SG In progress P16 Bacteria Diplorickettsia massiliensis 20B 2009 454_PE PRJNA86907 Published P17 Bacteria Enterobacter aerogenes EA1509E 2009 Sanger FO203355 Published P18 Bacteria Actinomyces grossensis 2012 SOLiD 454_PE 454_SG CAGY00000000Published P19 Bacteria Bacillus massiliosenegalensis 2012 SOLiD 454_PE 454_SG CAHJ00000000 Published P20 Bacteria Brevibacterium senegalensis -

Host-Microbe Relations: a Phylogenomics-Driven Bioinformatic Approach to the Characterization of Microbial DNA from Heterogeneous Sequence Data
Host-Microbe Relations: A Phylogenomics-Driven Bioinformatic Approach to the Characterization of Microbial DNA from Heterogeneous Sequence Data Timothy Patrick Driscoll Dissertation submitted to the faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy In Genetics, Bioinformatics, and Computational Biology Joseph J Gillespie David R Bevan Madhav V Marathe T M Murali May 1st, 2013 Blacksburg, Virginia Keywords: phylogenomics, genome-mining, host-microbe interactions, genomics, bioinformatics, symbiosis, bacteria, lateral gene transfer Copyright 2013 Host-Microbe Relations: A Phylogenomics-Driven Bioinformatic Approach to the Characterization of Microbial DNA from Heterogeneous Sequence Data Timothy Patrick Driscoll ABSTRACT Plants and animals are characterized by intimate, enduring, often indispensable, and always complex associations with microbes. Therefore, it should come as no surprise that when the genome of a eukaryote is sequenced, a medley of bacterial sequences are produced as well. These sequences can be highly informative about the interactions between the eukaryote and its bacterial cohorts; unfortunately, they often comprise a vanishingly small constituent within a heterogeneous mixture of microbial and host sequences. Genomic analyses typically avoid the bacterial sequences in order to obtain a genome sequence for the host. Metagenomic analysis typically avoid the host sequences in order to analyze community composition and functional diversity of the bacterial component. This dissertation describes the development of a novel approach at the intersection of genomics and metagenomics, aimed at the extraction and characterization of bacterial sequences from heterogeneous sequence data using phylogenomic and bioinformatic tools. To achieve this objective, three interoperable workflows were constructed as modular computational pipelines, with built-in checkpoints for periodic interpretation and refinement.