Detecting Phylogenetic Signals from Deep Roots of the Tree of Life
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Distribution of Tick-Borne Diseases in China Xian-Bo Wu1, Ren-Hua Na2, Shan-Shan Wei2, Jin-Song Zhu3 and Hong-Juan Peng2*
Wu et al. Parasites & Vectors 2013, 6:119 http://www.parasitesandvectors.com/content/6/1/119 REVIEW Open Access Distribution of tick-borne diseases in China Xian-Bo Wu1, Ren-Hua Na2, Shan-Shan Wei2, Jin-Song Zhu3 and Hong-Juan Peng2* Abstract As an important contributor to vector-borne diseases in China, in recent years, tick-borne diseases have attracted much attention because of their increasing incidence and consequent significant harm to livestock and human health. The most commonly observed human tick-borne diseases in China include Lyme borreliosis (known as Lyme disease in China), tick-borne encephalitis (known as Forest encephalitis in China), Crimean-Congo hemorrhagic fever (known as Xinjiang hemorrhagic fever in China), Q-fever, tularemia and North-Asia tick-borne spotted fever. In recent years, some emerging tick-borne diseases, such as human monocytic ehrlichiosis, human granulocytic anaplasmosis, and a novel bunyavirus infection, have been reported frequently in China. Other tick-borne diseases that are not as frequently reported in China include Colorado fever, oriental spotted fever and piroplasmosis. Detailed information regarding the history, characteristics, and current epidemic status of these human tick-borne diseases in China will be reviewed in this paper. It is clear that greater efforts in government management and research are required for the prevention, control, diagnosis, and treatment of tick-borne diseases, as well as for the control of ticks, in order to decrease the tick-borne disease burden in China. Keywords: Ticks, Tick-borne diseases, Epidemic, China Review (Table 1) [2,4]. Continuous reports of emerging tick-borne Ticks can carry and transmit viruses, bacteria, rickettsia, disease cases in Shandong, Henan, Hebei, Anhui, and spirochetes, protozoans, Chlamydia, Mycoplasma,Bartonia other provinces demonstrate the rise of these diseases bodies, and nematodes [1,2]. -

Bartonella Apis Sp. Nov., a Honey Bee Gut Symbiont of the Class Alphaproteobacteria
Serveur Academique´ Lausannois SERVAL serval.unil.ch Author Manuscript Faculty of Biology and Medicine Publication This paper has been peer-reviewed but does not include the final publisher proof-corrections or journal pagination. Published in final edited form as: Title: Bartonella apis sp. nov., a honey bee gut symbiont of the class Alphaproteobacteria. Authors: Keˇsnerov´aL, Moritz R, Engel P Journal: International journal of systematic and evolutionary microbiology Year: 2016 Jan Issue: 66 Volume: 1 Pages: 414-21 DOI: 10.1099/ijsem.0.000736 In the absence of a copyright statement, users should assume that standard copyright protection applies, unless the article contains an explicit statement to the contrary. In case of doubt, contact the journal publisher to verify the copyright status of an article. 1 Bartonella apis sp. nov., a honey bee gut symbiont of the 2 class Alphaproteobacteria 3 4 Lucie Kešnerová, Roxane Moritz, Philipp Engel* 5 6 Department of Fundamental Microbiology, University of Lausanne, CH-1015 7 Lausanne, Switzerland 8 9 Running title: Description of a bee gut symbiont 10 11 *Correspondence: 12 Prof. Philipp Engel 13 Department of Fundamental Microbiology 14 University of Lausanne, CH-1015 Lausanne, Switzerland 15 Tel.: +41 (0)21 692 56 12 16 e-mail: [email protected] 17 18 Category: New Taxa – Proteobacteria 19 Keywords: Apis mellifera; insect; Bartonella; gut microbiota; Alpha-1 20 21 Sequence deposition: The 16S rRNA gene sequences and protein-coding gene 22 sequences of the bacterial strains PEB0122T, PEB0149, PEB0150, BBC0104, and 23 BBC0108 from Apis mellifera, and the uncultured Rhizobiales bacterium from 24 Herpagnathos saltator are deposited in GenBank with accession numbers KP987849 25 – KP987886 and KT315729 – KT315734. -

Babela Massiliensis, a Representative of a Widespread Bacterial
Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae Isabelle Pagnier, Natalya Yutin, Olivier Croce, Kira S Makarova, Yuri I Wolf, Samia Benamar, Didier Raoult, Eugene V. Koonin, Bernard La Scola To cite this version: Isabelle Pagnier, Natalya Yutin, Olivier Croce, Kira S Makarova, Yuri I Wolf, et al.. Babela mas- siliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae. Biology Direct, BioMed Central, 2015, 10 (13), 10.1186/s13062-015-0043-z. hal-01217089 HAL Id: hal-01217089 https://hal-amu.archives-ouvertes.fr/hal-01217089 Submitted on 19 Oct 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Pagnier et al. Biology Direct (2015) 10:13 DOI 10.1186/s13062-015-0043-z RESEARCH Open Access Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae Isabelle Pagnier1, Natalya Yutin2, Olivier Croce1, Kira S Makarova2, Yuri I Wolf2, Samia Benamar1, Didier Raoult1, Eugene V Koonin2 and Bernard La Scola1* Abstract Background: Only a small fraction of bacteria and archaea that are identifiable by metagenomics can be grown on standard media. -

Characterization of the Interaction Between R. Conorii and Human
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 4-5-2018 Characterization of the Interaction Between R. Conorii and Human Host Vitronectin in Rickettsial Pathogenesis Abigail Inez Fish Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Bacteria Commons, Bacteriology Commons, Biology Commons, Immunology of Infectious Disease Commons, and the Pathogenic Microbiology Commons Recommended Citation Fish, Abigail Inez, "Characterization of the Interaction Between R. Conorii and Human Host Vitronectin in Rickettsial Pathogenesis" (2018). LSU Doctoral Dissertations. 4566. https://digitalcommons.lsu.edu/gradschool_dissertations/4566 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. CHARACTERIZATION OF THE INTERACTION BETWEEN R. CONORII AND HUMAN HOST VITRONECTIN IN RICKETTSIAL PATHOGENESIS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Interdepartmental Program in Biomedical and Veterinary Medical Sciences Through the Department of Pathobiological Sciences by Abigail Inez -

Alternative Hydrogen Uptake Pathways Suppress Methane Production In
bioRxiv preprint doi: https://doi.org/10.1101/486894; this version posted December 4, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 December 4, 2018 2 Alternative hydrogen uptake pathways 3 suppress methane production in ruminants 4 Chris Greening1 * #, Renae Geier2 #, Cecilia Wang3, Laura C. Woods1, Sergio E. 5 Morales3, Michael J. McDonald1, Rowena Rushton-Green3, Xochitl C. Morgan3, 6 Satoshi Koike4, Sinead C. Leahy5, William J. Kelly6, Isaac Cann2, Graeme T. 7 Attwood5, Gregory M. Cook3, Roderick I. Mackie2 * 8 9 1 Monash University, School of Biological Sciences, Clayton, VIC 3800, Australia 10 2 University of Illinois at Urbana-Champaign, Department of Animal Sciences and 11 Institute for Genomic Biology, Urbana, IL 61801, USA 12 3 University of Otago, Department of Microbiology and Immunology, Dunedin 9016, 13 New Zealand 14 4 Hokkaido University, Research Faculty of Agriculture, Sapporo, Japan 15 5 AgResearch Ltd., Grasslands Research Centre, Palmerston North 4410, New 16 Zealand. 17 6 Donvis Ltd., Palmerston North 4410, New Zealand. 18 19 # These authors contributed equally to this work. 20 21 * Correspondence can be addressed to: 22 23 Dr Chris Greening ([email protected]), School of Biological Sciences, 24 Monash University, Clayton, VIC 3800, Australia 25 Prof Roderick Mackie ([email protected]), Department of Animal Sciences, 26 Urbana, IL 61801, USA 27 bioRxiv preprint doi: https://doi.org/10.1101/486894; this version posted December 4, 2018. -

Sanguineus Bacterial Communities Rhipicephalus
Composition and Seasonal Variation of Rhipicephalus turanicus and Rhipicephalus sanguineus Bacterial Communities Itai Lalzar, Shimon Harrus, Kosta Y. Mumcuoglu and Yuval Gottlieb Appl. Environ. Microbiol. 2012, 78(12):4110. DOI: 10.1128/AEM.00323-12. Published Ahead of Print 30 March 2012. Downloaded from Updated information and services can be found at: http://aem.asm.org/content/78/12/4110 These include: SUPPLEMENTAL MATERIAL Supplemental material http://aem.asm.org/ REFERENCES This article cites 44 articles, 17 of which can be accessed free at: http://aem.asm.org/content/78/12/4110#ref-list-1 CONTENT ALERTS Receive: RSS Feeds, eTOCs, free email alerts (when new articles cite this article), more» on June 10, 2013 by guest Information about commercial reprint orders: http://journals.asm.org/site/misc/reprints.xhtml To subscribe to to another ASM Journal go to: http://journals.asm.org/site/subscriptions/ Composition and Seasonal Variation of Rhipicephalus turanicus and Rhipicephalus sanguineus Bacterial Communities Itai Lalzar,a Shimon Harrus,a Kosta Y. Mumcuoglu,b and Yuval Gottlieba Koret School of Veterinary Medicine, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, Rehovot, Israel,a and Department of Microbiology and Molecular Genetics, The Kuvin Center for the Study of Infectious and Tropical Diseases, Hadassah Medical School, The Institute for Medical Research Israel-Canada, The Hebrew University of Jerusalem, Jerusalem, Israelb A 16S rRNA gene approach, including 454 pyrosequencing and quantitative PCR (qPCR), was used to describe the bacterial com- munity in Rhipicephalus turanicus and to evaluate the dynamics of key bacterial tenants of adult ticks during the active questing season. -
LC-Locus Alignment Sites Distance, Number of Nodes Supplementary
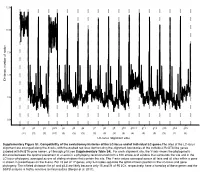
12.5 10.0 7.5 5.0 Distance, number of nodes 2.5 0.0 g1 g2 g3 g3.5 g4 g5 g6 g7 g8 g9 g10 g10.1 g11 g12 g13 g14 g15 (11) (3) (3) (10) (3) (3) (3) (3) (3) (3) (3) (4) (3) (3) (3) (2) (3) LC-locus alignment sites Supplementary Figure S1. Compatibility of the evolutionary histories of the LC-locus and of individual LC genes.The sites of the LC-locus alignment are arranged along the X-axis, with the dashed red lines demarcating the alignment boundaries of the individual RcGTA-like genes (labeled with RcGTA gene names, g1 through g15; see Supplementary Table S4). For each alignment site, the Y-axis shows the phylogenetic distance between the optimal placement of a taxon in a phylogeny reconstructed from a 100 amino-acid window that surrounds the site and in the LC-locus phylogeny, averaged across all sliding windows that contain the site. The Y-axis values averaged across all taxa and all sites within a gene is shown in parentheses on the X-axis. For 15 out of 17 genes, only 2-4 nodes separate the optimal taxon position in the LC-locus and gene phylogeny. The inflated distances for g1 and g3.5 are likely because only 15 and 21 of 95 LCs, respectively, have a homolog of these genes and the SSPB analysis is highly sensitive to missing data (Berger et al. 2011). a. Bacteria Unassigned Thermotogae Tenericutes Synergistetes Spirochaetes Proteobacteria ylum Planctomycetes h p Firmicutes Deferribacteres Cyanobacteria Chloroflexi Bacteroidetes Actinobacteria Acidobacteria 1(11,750) 2(1,750) 3(2,538) 4(168) 5(51) 6(54) 7(43) 8(32) 9(26) 10(40) 11(33) 12(198) 13(173) 14(101) 15(98) 16(43) 17(114) Number of rcc01682−rcc01698 homologs in a cluster b. -

Genome Project Reveals a Putative Rickettsial Endosymbiont
GBE Bacterial DNA Sifted from the Trichoplax adhaerens (Animalia: Placozoa) Genome Project Reveals a Putative Rickettsial Endosymbiont Timothy Driscoll1,y, Joseph J. Gillespie1,2,*,y, Eric K. Nordberg1,AbduF.Azad2, and Bruno W. Sobral1,3 1Virginia Bioinformatics Institute at Virginia Polytechnic Institute and State University 2Department of Microbiology and Immunology, University of Maryland School of Medicine 3Present address: Nestle´ Institute of Health Sciences SA, Campus EPFL, Quartier de L’innovation, Lausanne, Switzerland *Corresponding author: E-mail: [email protected]. yThese authors contributed equally to this work. Accepted: March 1, 2013 Abstract Eukaryotic genome sequencing projects often yield bacterial DNA sequences, data typically considered as microbial contamination. However, these sequences may also indicate either symbiont genes or lateral gene transfer (LGT) to host genomes. These bacterial sequences can provide clues about eukaryote–microbe interactions. Here, we used the genome of the primitive animal Trichoplax adhaerens (Metazoa: Placozoa), which is known to harbor an uncharacterized Gram-negative endosymbiont, to search for the presence of bacterial DNA sequences. Bioinformatic and phylogenomic analyses of extracted data from the genome assembly (181 bacterial coding sequences [CDS]) and trace read archive (16S rDNA) revealed a dominant proteobacterial profile strongly skewed to Rickettsiales (Alphaproteobacteria) genomes. By way of phylogenetic analysis of 16S rDNA and 113 proteins conserved across proteobacterial genomes, as well as identification of 27 rickettsial signature genes, we propose a Rickettsiales endosymbiont of T. adhaerens (RETA). The majority (93%) of the identified bacterial CDS belongs to small scaffolds containing prokaryotic-like genes; however, 12 CDS were identified on large scaffolds comprised of eukaryotic-like genes, suggesting that T. -

Detection and Partial Molecular Characterization of Rickettsia and Bartonella from Southern African Bat Species
Detection and partial molecular characterization of Rickettsia and Bartonella from southern African bat species by Tjale Mabotse Augustine (29685690) Submitted in partial fulfillment of the requirements for the degree MAGISTER SCIENTIAE (MICROBIOLOGY) in the Department of Microbiology and Plant Pathology Faculty of Natural and Agricultural Sciences University of Pretoria Pretoria, South Africa Supervisor: Dr Wanda Markotter Co-supervisors: Prof Louis H. Nel Dr Jacqueline Weyer May, 2012 I declare that the thesis, which I hereby submit for the degree MSc (Microbiology) at the University of Pretoria, South Africa, is my own work and has not been submitted by me for a degree at another university ________________________________ Tjale Mabotse Augustine i Acknowledgements I would like send my sincere gratitude to the following people: Dr Wanda Markotter (University of Pretoria), Dr Jacqueline Weyer (National Institute for Communicable Diseases-National Health Laboratory Service) and Prof Louis H Nel (University of Pretoria) for their supervision and guidance during the project. Dr Jacqueline Weyer (Centre for Zoonotic and Emerging diseases (Previously Special Pathogens Unit), National Institute for Communicable Diseases (National Heath Laboratory Service), for providing the positive control DNA for Rickettsia and Dr Jenny Rossouw (Special Bacterial Pathogens Reference Unit, National Institute for Communicable Diseases-National Health Laboratory Service), for providing the positive control DNA for Bartonella. Dr Teresa Kearney (Ditsong Museum of Natural Science), Gauteng and Northern Region Bat Interest Group, Kwa-Zulu Natal Bat Interest Group, Prof Ara Monadjem (University of Swaziland), Werner Marias (University of Johannesburg), Dr Francois du Rand (University of Johannesburg) and Prof David Jacobs (University of Cape Town) for collection of blood samples. -

Ohio Department of Health, Bureau of Infectious Diseases Disease Name Class A, Requires Immediate Phone Call to Local Health
Ohio Department of Health, Bureau of Infectious Diseases Reporting specifics for select diseases reportable by ELR Class A, requires immediate phone Susceptibilities specimen type Reportable test name (can change if Disease Name other specifics+ call to local health required* specifics~ state/federal case definition or department reporting requirements change) Culture independent diagnostic tests' (CIDT), like BioFire panel or BD MAX, E. histolytica Stain specimen = stool, bile results should be sent as E. histolytica DNA fluid, duodenal fluid, 260373001^DETECTED^SCT with E. histolytica Antigen Amebiasis (Entamoeba histolytica) No No tissue large intestine, disease/organism-specific DNA LOINC E. histolytica Antibody tissue small intestine codes OR a generic CIDT-LOINC code E. histolytica IgM with organism-specific DNA SNOMED E. histolytica IgG codes E. histolytica Total Antibody Ova and Parasite Anthrax Antibody Anthrax Antigen Anthrax EITB Acute Anthrax EITB Convalescent Anthrax Yes No Culture ELISA PCR Stain/microscopy Stain/spore ID Eastern Equine Encephalitis virus Antibody Eastern Equine Encephalitis virus IgG Antibody Eastern Equine Encephalitis virus IgM Arboviral neuroinvasive and non- Eastern Equine Encephalitis virus RNA neuroinvasive disease: Eastern equine California serogroup virus Antibody encephalitis virus disease; LaCrosse Equivocal results are accepted for all California serogroup virus IgG Antibody virus disease (other California arborviral diseases; California serogroup virus IgM Antibody specimen = blood, serum, serogroup -

Supplemental Material S1.Pdf
Phylogeny of Selenophosphate synthetases (SPS) Supplementary Material S1 ! SelD in prokaryotes! ! ! SelD gene finding in sequenced prokaryotes! We downloaded a total of 8263 prokaryotic genomes from NCBI (see Supplementary Material S7). We scanned them with the program selenoprofiles (Mariotti 2010, http:// big.crg.cat/services/selenoprofiles) using two SPS-family profiles, one prokaryotic (seld) and one mixed eukaryotic-prokaryotic (SPS). Selenoprofiles removes overlapping predictions from different profiles, keeping only the prediction from the profile that seems closer to the candidate sequence. As expected, the great majority of output predictions in prokaryotic genomes were from the seld profile. We will refer to the prokaryotic SPS/SelD !genes as SelD, following the most common nomenclature in literature.! To be able to inspect results by hand, and also to focus on good-quality genomes, we considered a reduced set of species. We took the prok_reference_genomes.txt list from ftp://ftp.ncbi.nlm.nih.gov/genomes/GENOME_REPORTS/, which NCBI claims to be a "small curated subset of really good and scientifically important prokaryotic genomes". We named this the prokaryotic reference set (223 species - see Supplementary Material S8). We manually curated most of the analysis in this set, while we kept automatized the !analysis on the full set.! We detected SelD proteins in 58 genomes (26.0%) in the prokaryotic reference set (figure 1 in main paper), which become 2805 (33.9%) when considering the prokaryotic full set (figure SM1.1). The difference in proportion between the two sets is due largely to the presence of genomes of very close strains in the full set, which we consider redundant. -

Expansion of Tick-Borne Rickettsioses in the World
microorganisms Review Expansion of Tick-Borne Rickettsioses in the World Mariusz Piotrowski * and Anna Rymaszewska Institute of Biology, University of Szczecin, 70-453 Szczecin, Poland; [email protected] * Correspondence: [email protected] Received: 24 September 2020; Accepted: 25 November 2020; Published: 30 November 2020 Abstract: Tick-borne rickettsioses are caused by obligate intracellular bacteria belonging to the spotted fever group of the genus Rickettsia. These infections are among the oldest known diseases transmitted by vectors. In the last three decades there has been a rapid increase in the recognition of this disease complex. This unusual expansion of information was mainly caused by the development of molecular diagnostic techniques that have facilitated the identification of new and previously recognized rickettsiae. A lot of currently known bacteria of the genus Rickettsia have been considered nonpathogenic for years, and moreover, many new species have been identified with unknown pathogenicity. The genus Rickettsia is distributed all over the world. Many Rickettsia species are present on several continents. The geographical distribution of rickettsiae is related to their vectors. New cases of rickettsioses and new locations, where the presence of these bacteria is recognized, are still being identified. The variety and rapid evolution of the distribution and density of ticks and diseases which they transmit shows us the scale of the problem. This review article presents a comparison of the current understanding of the geographic distribution of pathogenic Rickettsia species to that of the beginning of the century. Keywords: Tick-borne rickettsioses; Tick-borne diseases; Rickettsiales 1. Introduction Tick-borne rickettsioses are caused by obligate intracellular Gram-negative bacteria belonging to the spotted fever group (SFG) of the genus Rickettsia.