Babela Massiliensis, a Representative of A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metaproteogenomic Insights Beyond Bacterial Response to Naphthalene
ORIGINAL ARTICLE ISME Journal – Original article Metaproteogenomic insights beyond bacterial response to 5 naphthalene exposure and bio-stimulation María-Eugenia Guazzaroni, Florian-Alexander Herbst, Iván Lores, Javier Tamames, Ana Isabel Peláez, Nieves López-Cortés, María Alcaide, Mercedes V. del Pozo, José María Vieites, Martin von Bergen, José Luis R. Gallego, Rafael Bargiela, Arantxa López-López, Dietmar H. Pieper, Ramón Rosselló-Móra, Jesús Sánchez, Jana Seifert and Manuel Ferrer 10 Supporting Online Material includes Text (Supporting Materials and Methods) Tables S1 to S9 Figures S1 to S7 1 SUPPORTING TEXT Supporting Materials and Methods Soil characterisation Soil pH was measured in a suspension of soil and water (1:2.5) with a glass electrode, and 5 electrical conductivity was measured in the same extract (diluted 1:5). Primary soil characteristics were determined using standard techniques, such as dichromate oxidation (organic matter content), the Kjeldahl method (nitrogen content), the Olsen method (phosphorus content) and a Bernard calcimeter (carbonate content). The Bouyoucos Densimetry method was used to establish textural data. Exchangeable cations (Ca, Mg, K and 10 Na) extracted with 1 M NH 4Cl and exchangeable aluminium extracted with 1 M KCl were determined using atomic absorption/emission spectrophotometry with an AA200 PerkinElmer analyser. The effective cation exchange capacity (ECEC) was calculated as the sum of the values of the last two measurements (sum of the exchangeable cations and the exchangeable Al). Analyses were performed immediately after sampling. 15 Hydrocarbon analysis Extraction (5 g of sample N and Nbs) was performed with dichloromethane:acetone (1:1) using a Soxtherm extraction apparatus (Gerhardt GmbH & Co. -

Babela Massiliensis, a Representative of a Widespread Bacterial
Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae Isabelle Pagnier, Natalya Yutin, Olivier Croce, Kira S Makarova, Yuri I Wolf, Samia Benamar, Didier Raoult, Eugene V. Koonin, Bernard La Scola To cite this version: Isabelle Pagnier, Natalya Yutin, Olivier Croce, Kira S Makarova, Yuri I Wolf, et al.. Babela mas- siliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae. Biology Direct, BioMed Central, 2015, 10 (13), 10.1186/s13062-015-0043-z. hal-01217089 HAL Id: hal-01217089 https://hal-amu.archives-ouvertes.fr/hal-01217089 Submitted on 19 Oct 2015 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Pagnier et al. Biology Direct (2015) 10:13 DOI 10.1186/s13062-015-0043-z RESEARCH Open Access Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae Isabelle Pagnier1, Natalya Yutin2, Olivier Croce1, Kira S Makarova2, Yuri I Wolf2, Samia Benamar1, Didier Raoult1, Eugene V Koonin2 and Bernard La Scola1* Abstract Background: Only a small fraction of bacteria and archaea that are identifiable by metagenomics can be grown on standard media. -

BD-CS-057, REV 0 | AUGUST 2017 | Page 1
EXPLIFY RESPIRATORY PATHOGENS BY NEXT GENERATION SEQUENCING Limitations Negative results do not rule out viral, bacterial, or fungal infections. Targeted, PCR-based tests are generally more sensitive and are preferred when specific pathogens are suspected, especially for DNA viruses (Adenovirus, CMV, HHV6, HSV, and VZV), mycobacteria, and fungi. The analytical sensitivity of this test depends on the cellularity of the sample and the concentration of all microbes present. Analytical sensitivity is assessed using Internal Controls that are added to each sample. Sequencing data for Internal Controls is quantified. Samples with Internal Control values below the validated minimum may have reduced analytical sensitivity or contain inhibitors and are reported as ‘Reduced Analytical Sensitivity’. Additional respiratory pathogens to those reported cannot be excluded in samples with ‘Reduced Analytical Sensitivity’. Due to the complexity of next generation sequencing methodologies, there may be a risk of false-positive results. Contamination with organisms from the upper respiratory tract during specimen collection can also occur. The detection of viral, bacterial, and fungal nucleic acid does not imply organisms causing invasive infection. Results from this test need to be interpreted in conjunction with the clinical history, results of other laboratory tests, epidemiologic information, and other available data. Confirmation of positive results by an alternate method may be indicated in select cases. Validated Organisms BACTERIA Achromobacter -

Evolutionary Origin of Insect–Wolbachia Nutritional Mutualism
Evolutionary origin of insect–Wolbachia nutritional mutualism Naruo Nikoha,1, Takahiro Hosokawab,1, Minoru Moriyamab,1, Kenshiro Oshimac, Masahira Hattoric, and Takema Fukatsub,2 aDepartment of Liberal Arts, The Open University of Japan, Chiba 261-8586, Japan; bBioproduction Research Institute, National Institute of Advanced Industrial Science and Technology, Tsukuba 305-8566, Japan; and cCenter for Omics and Bioinformatics, Graduate School of Frontier Sciences, University of Tokyo, Kashiwa 277-8561, Japan Edited by Nancy A. Moran, University of Texas at Austin, Austin, TX, and approved June 3, 2014 (received for review May 20, 2014) Obligate insect–bacterium nutritional mutualism is among the insects, generally conferring negative fitness consequences to most sophisticated forms of symbiosis, wherein the host and the their hosts and often causing hosts’ reproductive aberrations to symbiont are integrated into a coherent biological entity and un- enhance their own transmission in a selfish manner (7, 8). Re- able to survive without the partnership. Originally, however, such cently, however, a Wolbachia strain associated with the bedbug obligate symbiotic bacteria must have been derived from free-living Cimex lectularius,designatedaswCle, was shown to be es- bacteria. How highly specialized obligate mutualisms have arisen sential for normal growth and reproduction of the blood- from less specialized associations is of interest. Here we address this sucking insect host via provisioning of B vitamins (9). Hence, it –Wolbachia evolutionary -
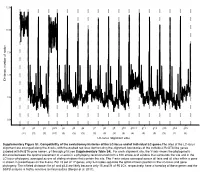
LC-Locus Alignment Sites Distance, Number of Nodes Supplementary
12.5 10.0 7.5 5.0 Distance, number of nodes 2.5 0.0 g1 g2 g3 g3.5 g4 g5 g6 g7 g8 g9 g10 g10.1 g11 g12 g13 g14 g15 (11) (3) (3) (10) (3) (3) (3) (3) (3) (3) (3) (4) (3) (3) (3) (2) (3) LC-locus alignment sites Supplementary Figure S1. Compatibility of the evolutionary histories of the LC-locus and of individual LC genes.The sites of the LC-locus alignment are arranged along the X-axis, with the dashed red lines demarcating the alignment boundaries of the individual RcGTA-like genes (labeled with RcGTA gene names, g1 through g15; see Supplementary Table S4). For each alignment site, the Y-axis shows the phylogenetic distance between the optimal placement of a taxon in a phylogeny reconstructed from a 100 amino-acid window that surrounds the site and in the LC-locus phylogeny, averaged across all sliding windows that contain the site. The Y-axis values averaged across all taxa and all sites within a gene is shown in parentheses on the X-axis. For 15 out of 17 genes, only 2-4 nodes separate the optimal taxon position in the LC-locus and gene phylogeny. The inflated distances for g1 and g3.5 are likely because only 15 and 21 of 95 LCs, respectively, have a homolog of these genes and the SSPB analysis is highly sensitive to missing data (Berger et al. 2011). a. Bacteria Unassigned Thermotogae Tenericutes Synergistetes Spirochaetes Proteobacteria ylum Planctomycetes h p Firmicutes Deferribacteres Cyanobacteria Chloroflexi Bacteroidetes Actinobacteria Acidobacteria 1(11,750) 2(1,750) 3(2,538) 4(168) 5(51) 6(54) 7(43) 8(32) 9(26) 10(40) 11(33) 12(198) 13(173) 14(101) 15(98) 16(43) 17(114) Number of rcc01682−rcc01698 homologs in a cluster b. -

Legionella Shows a Diverse Secondary Metabolism Dependent on a Broad Spectrum Sfp-Type Phosphopantetheinyl Transferase
Legionella shows a diverse secondary metabolism dependent on a broad spectrum Sfp-type phosphopantetheinyl transferase Nicholas J. Tobias1, Tilman Ahrendt1, Ursula Schell2, Melissa Miltenberger1, Hubert Hilbi2,3 and Helge B. Bode1,4 1 Fachbereich Biowissenschaften, Merck Stiftungsprofessur fu¨r Molekulare Biotechnologie, Goethe Universita¨t, Frankfurt am Main, Germany 2 Max von Pettenkofer Institute, Ludwig-Maximilians-Universita¨tMu¨nchen, Munich, Germany 3 Institute of Medical Microbiology, University of Zu¨rich, Zu¨rich, Switzerland 4 Buchmann Institute for Molecular Life Sciences, Goethe Universita¨t, Frankfurt am Main, Germany ABSTRACT Several members of the genus Legionella cause Legionnaires’ disease, a potentially debilitating form of pneumonia. Studies frequently focus on the abundant number of virulence factors present in this genus. However, what is often overlooked is the role of secondary metabolites from Legionella. Following whole genome sequencing, we assembled and annotated the Legionella parisiensis DSM 19216 genome. Together with 14 other members of the Legionella, we performed comparative genomics and analysed the secondary metabolite potential of each strain. We found that Legionella contains a huge variety of biosynthetic gene clusters (BGCs) that are potentially making a significant number of novel natural products with undefined function. Surprisingly, only a single Sfp-like phosphopantetheinyl transferase is found in all Legionella strains analyzed that might be responsible for the activation of all carrier proteins in primary (fatty acid biosynthesis) and secondary metabolism (polyketide and non-ribosomal peptide synthesis). Using conserved active site motifs, we predict Submitted 29 June 2016 some novel compounds that are probably involved in cell-cell communication, Accepted 25 October 2016 Published 24 November 2016 differing to known communication systems. -

Ohio Department of Health, Bureau of Infectious Diseases Disease Name Class A, Requires Immediate Phone Call to Local Health
Ohio Department of Health, Bureau of Infectious Diseases Reporting specifics for select diseases reportable by ELR Class A, requires immediate phone Susceptibilities specimen type Reportable test name (can change if Disease Name other specifics+ call to local health required* specifics~ state/federal case definition or department reporting requirements change) Culture independent diagnostic tests' (CIDT), like BioFire panel or BD MAX, E. histolytica Stain specimen = stool, bile results should be sent as E. histolytica DNA fluid, duodenal fluid, 260373001^DETECTED^SCT with E. histolytica Antigen Amebiasis (Entamoeba histolytica) No No tissue large intestine, disease/organism-specific DNA LOINC E. histolytica Antibody tissue small intestine codes OR a generic CIDT-LOINC code E. histolytica IgM with organism-specific DNA SNOMED E. histolytica IgG codes E. histolytica Total Antibody Ova and Parasite Anthrax Antibody Anthrax Antigen Anthrax EITB Acute Anthrax EITB Convalescent Anthrax Yes No Culture ELISA PCR Stain/microscopy Stain/spore ID Eastern Equine Encephalitis virus Antibody Eastern Equine Encephalitis virus IgG Antibody Eastern Equine Encephalitis virus IgM Arboviral neuroinvasive and non- Eastern Equine Encephalitis virus RNA neuroinvasive disease: Eastern equine California serogroup virus Antibody encephalitis virus disease; LaCrosse Equivocal results are accepted for all California serogroup virus IgG Antibody virus disease (other California arborviral diseases; California serogroup virus IgM Antibody specimen = blood, serum, serogroup -

Gene Gain and Loss Events in Rickettsia and Orientia Species Kalliopi Georgiades1,2, Vicky Merhej1, Khalid El Karkouri1, Didier Raoult1, Pierre Pontarotti2*
Georgiades et al. Biology Direct 2011, 6:6 http://www.biology-direct.com/content/6/1/6 RESEARCH Open Access Gene gain and loss events in Rickettsia and Orientia species Kalliopi Georgiades1,2, Vicky Merhej1, Khalid El Karkouri1, Didier Raoult1, Pierre Pontarotti2* Abstract Background: Genome degradation is an ongoing process in all members of the Rickettsiales order, which makes these bacterial species an excellent model for studying reductive evolution through interspecies variation in genome size and gene content. In this study, we evaluated the degree to which gene loss shaped the content of some Rickettsiales genomes. We shed light on the role played by horizontal gene transfers in the genome evolution of Rickettsiales. Results: Our phylogenomic tree, based on whole-genome content, presented a topology distinct from that of the whole core gene concatenated phylogenetic tree, suggesting that the gene repertoires involved have different evolutionary histories. Indeed, we present evidence for 3 possible horizontal gene transfer events from various organisms to Orientia and 6 to Rickettsia spp., while we also identified 3 possible horizontal gene transfer events from Rickettsia and Orientia to other bacteria. We found 17 putative genes in Rickettsia spp. that are probably the result of de novo gene creation; 2 of these genes appear to be functional. On the basis of these results, we were able to reconstruct the gene repertoires of “proto-Rickettsiales” and “proto-Rickettsiaceae”, which correspond to the ancestors of Rickettsiales and Rickettsiaceae, respectively. Finally, we found that 2,135 genes were lost during the evolution of the Rickettsiaceae to an intracellular lifestyle. Conclusions: Our phylogenetic analysis allowed us to track the gene gain and loss events occurring in bacterial genomes during their evolution from a free-living to an intracellular lifestyle. -

The Risk to Human Health from Free-Living Amoebae Interaction with Legionella in Drinking and Recycled Water Systems
THE RISK TO HUMAN HEALTH FROM FREE-LIVING AMOEBAE INTERACTION WITH LEGIONELLA IN DRINKING AND RECYCLED WATER SYSTEMS Dissertation submitted by JACQUELINE MARIE THOMAS BACHELOR OF SCIENCE (HONOURS) AND BACHELOR OF ARTS, UNSW In partial fulfillment of the requirements for the award of DOCTOR OF PHILOSOPHY in ENVIRONMENTAL ENGINEERING SCHOOL OF CIVIL AND ENVIRONMENTAL ENGINEERING FACULTY OF ENGINEERING MAY 2012 SUPERVISORS Professor Nicholas Ashbolt Office of Research and Development United States Environmental Protection Agency Cincinnati, Ohio USA and School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Professor Richard Stuetz School of Civil and Environmental Engineering Faculty of Engineering The University of New South Wales Sydney, Australia Doctor Torsten Thomas School of Biotechnology and Biomolecular Sciences Faculty of Science The University of New South Wales Sydney, Australia ORIGINALITY STATEMENT '1 hereby declare that this submission is my own work and to the best of my knowledge it contains no materials previously published or written by another person, or substantial proportions of material which have been accepted for the award of any other degree or diploma at UNSW or any other educational institution, except where due acknowledgement is made in the thesis. Any contribution made to the research by others, with whom 1 have worked at UNSW or elsewhere, is explicitly acknowledged in the thesis. I also declare that the intellectual content of this thesis is the product of my own work, except to the extent that assistance from others in the project's design and conception or in style, presentation and linguistic expression is acknowledged.' Signed ~ ............................ -

High Prevalence of Rickettsia Africae Variants in Amblyomma Variegatum Ticks from Domestic Mammals in Rural Western Kenya: Implications for Human Health
n Maina, A. N. et al. (2014) High prevalence of Rickettsia africae variants in Amblyomma variegatum ticks from domestic mammals in rural western Kenya: implications for human health. Vector-Borne and Zoonotic Diseases, 14 (10). pp. 693-702. ISSN 1530-3667 Copyright © 2014 The Authors http://eprints.gla.ac.uk/99480/ Deposited on: 17 November 2014 Enlighten – Research publications by members of the University of Glasgow http://eprints.gla.ac.uk VECTOR-BORNE AND ZOONOTIC DISEASES Volume 14, Number 10, 2014 ORIGINAL ARTICLES ª Mary Ann Liebert, Inc. DOI: 10.1089/vbz.2014.1578 High Prevalence of Rickettsia africae Variants in Amblyomma variegatum Ticks from Domestic Mammals in Rural Western Kenya: Implications for Human Health Alice N. Maina,1–3 Ju Jiang,3 Sylvia A. Omulo,2 Sally J. Cutler,4 Fredrick Ade,2 Eric Ogola,2 Daniel R. Feikin,5 M. Kariuki Njenga,5 Sarah Cleaveland,6 Solomon Mpoke,1,2 Zipporah Ng’ang’a,1 Robert F. Breiman,5 Darryn L. Knobel,2,7 and Allen L. Richards3 Abstract Tick-borne spotted fever group (SFG) rickettsioses are emerging human diseases caused by obligate intracel- lular Gram-negative bacteria of the genus Rickettsia. Despite being important causes of systemic febrile illnesses in travelers returning from sub-Saharan Africa, little is known about the reservoir hosts of these pathogens. We conducted surveys for rickettsiae in domestic animals and ticks in a rural setting in western Kenya. Of the 100 serum specimens tested from each species of domestic ruminant 43% of goats, 23% of sheep, and 1% of cattle had immunoglobulin G (IgG) antibodies to the SFG rickettsiae. -

18167638.Pdf
REVIEW ARTICLE Amoebal pathogens as emerging causal agents of pneumonia Fred´ eric´ Lamoth1 & Gilbert Greub1,2 1Infectious Diseases Service, University of Lausanne, Lausanne, Switzerland; and 2Center for Research on Intracellular Bacteria, Institute of Microbiology, University Hospital Center and University of Lausanne, Lausanne, Switzerland Correspondence: Gilbert Greub, Center for Abstract Research on Intracellular Bacteria, Institute of Microbiology, University Hospital Center and Despite using modern microbiological diagnostic approaches, the aetiological University of Lausanne, Rue du Bugnon 46, agents of pneumonia remain unidentified in about 50% of cases. Some bacteria 1011 Lausanne, Switzerland. Tel.: 141 21 that grow poorly or not at all in axenic media used in routine clinical bacteriology 31449 79; fax: 141 21 31440 60; e-mail: laboratory but which can develop inside amoebae may be the agents of these lower [email protected] respiratory tract infections (RTIs) of unexplained aetiology. Such amoebae- resisting bacteria, which coevolved with amoebae to resist their microbicidal Received 24 September 2009; revised 30 machinery, may have developed virulence traits that help them survive within November 2009; accepted 2 December 2009. human macrophages, i.e. the first line of innate immune defence in the lung. We Final version published online 22 January 2010. review here the current evidence for the emerging pathogenic role of various DOI:10.1111/j.1574-6976.2009.00207.x amoebae-resisting microorganisms as agents of RTIs in humans. Specifically, we discuss the emerging pathogenic roles of Legionella-like amoebal pathogens, novel Editor: Colin Berry Chlamydiae (Parachlamydia acanthamoebae, Simkania negevensis), waterborne mycobacteria and Bradyrhizobiaceae (Bosea and Afipia spp.). Keywords free-living amoebae; amoebae-resisting bacteria; Legionella; Chlamydia-like bacteria; mycobacteria; pneumonia. -

Mariem Joan Wasan Oloroso
Interactions between Arcobacter butzleri and free-living protozoa in the context of sewage & wastewater treatment by Mariem Joan Wasan Oloroso A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Environmental Health Sciences School of Public Health University of Alberta © Mariem Joan Wasan Oloroso, 2021 Abstract Water reuse is increasingly becoming implemented as a sustainable water management strategy in areas around the world facing freshwater shortages and nutrient discharge limits. However, there are a host of biological hazards that must be assessed prior to and following the introduction of water reuse schemes. Members of the genus Arcobacter are close relatives to the well-known foodborne campylobacter pathogens and are increasingly being recognized as emerging human pathogens of concern. Arcobacters are prevalent in numerous water environments due to their ability to survive in a wide range of conditions. They are particularly abundant in raw sewage and are able to survive wastewater treatment and disinfection processes, which marks this genus as a potential pathogen of concern for water quality. Because the low levels of Arcobacter excreted by humans do not correlate with the high levels of Arcobacter spp. present in raw sewage, it was hypothesised that other microorganisms in sewage may amplify the growth of Arcobacter species. There is evidence that Arcobacter spp. survive both within and on the surface of free-living protozoa (FLP). As such, this thesis investigated the idea that Arcobacter spp. may be growing within free-living protozoa also prevalent in raw sewage and providing them with protection during treatment and disinfection processes.