GLUCOSE ISOMERASE from an ARTHROBACTER SPECIES By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

(12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk Et Al
US 2014O155567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk et al. (43) Pub. Date: Jun. 5, 2014 (54) MICROORGANISMS AND METHODS FOR (60) Provisional application No. 61/331,812, filed on May THE BIOSYNTHESIS OF BUTADENE 5, 2010. (71) Applicant: Genomatica, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Mark J. Burk, San Diego, CA (US); (51) Int. Cl. Anthony P. Burgard, Bellefonte, PA CI2P 5/02 (2006.01) (US); Jun Sun, San Diego, CA (US); CSF 36/06 (2006.01) Robin E. Osterhout, San Diego, CA CD7C II/6 (2006.01) (US); Priti Pharkya, San Diego, CA (52) U.S. Cl. (US) CPC ................. CI2P5/026 (2013.01); C07C II/I6 (2013.01); C08F 136/06 (2013.01) (73) Assignee: Genomatica, Inc., San Diego, CA (US) USPC ... 526/335; 435/252.3:435/167; 435/254.2: (21) Appl. No.: 14/059,131 435/254.11: 435/252.33: 435/254.21:585/16 (22) Filed: Oct. 21, 2013 (57) ABSTRACT O O The invention provides non-naturally occurring microbial Related U.S. Application Data organisms having a butadiene pathway. The invention addi (63) Continuation of application No. 13/101,046, filed on tionally provides methods of using Such organisms to produce May 4, 2011, now Pat. No. 8,580,543. butadiene. Patent Application Publication Jun. 5, 2014 Sheet 1 of 4 US 2014/O155567 A1 ?ueudos!SMS |?un61– Patent Application Publication Jun. 5, 2014 Sheet 2 of 4 US 2014/O155567 A1 VOJ OO O Z?un61– Patent Application Publication US 2014/O155567 A1 {}}} Hººso Patent Application Publication Jun. -

Preclinical Evaluation of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma by Andrea Shergalis
Preclinical Evaluation of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma By Andrea Shergalis A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Medicinal Chemistry) in the University of Michigan 2020 Doctoral Committee: Professor Nouri Neamati, Chair Professor George A. Garcia Professor Peter J. H. Scott Professor Shaomeng Wang Andrea G. Shergalis [email protected] ORCID 0000-0002-1155-1583 © Andrea Shergalis 2020 All Rights Reserved ACKNOWLEDGEMENTS So many people have been involved in bringing this project to life and making this dissertation possible. First, I want to thank my advisor, Prof. Nouri Neamati, for his guidance, encouragement, and patience. Prof. Neamati instilled an enthusiasm in me for science and drug discovery, while allowing me the space to independently explore complex biochemical problems, and I am grateful for his kind and patient mentorship. I also thank my committee members, Profs. George Garcia, Peter Scott, and Shaomeng Wang, for their patience, guidance, and support throughout my graduate career. I am thankful to them for taking time to meet with me and have thoughtful conversations about medicinal chemistry and science in general. From the Neamati lab, I would like to thank so many. First and foremost, I have to thank Shuzo Tamara for being an incredible, kind, and patient teacher and mentor. Shuzo is one of the hardest workers I know. In addition to a strong work ethic, he taught me pretty much everything I know and laid the foundation for the article published as Chapter 3 of this dissertation. The work published in this dissertation really began with the initial identification of PDI as a target by Shili Xu, and I am grateful for his advice and guidance (from afar!). -

Pyruvate-Phosphate Dikinase of Oxymonads and Parabasalia and the Evolution of Pyrophosphate-Dependent Glycolysis in Anaerobic Eukaryotes† Claudio H
EUKARYOTIC CELL, Jan. 2006, p. 148–154 Vol. 5, No. 1 1535-9778/06/$08.00ϩ0 doi:10.1128/EC.5.1.148–154.2006 Copyright © 2006, American Society for Microbiology. All Rights Reserved. Pyruvate-Phosphate Dikinase of Oxymonads and Parabasalia and the Evolution of Pyrophosphate-Dependent Glycolysis in Anaerobic Eukaryotes† Claudio H. Slamovits and Patrick J. Keeling* Canadian Institute for Advanced Research, Botany Department, University of British Columbia, 3529-6270 University Boulevard, Vancouver, British Columbia V6T 1Z4, Canada Received 29 September 2005/Accepted 8 November 2005 In pyrophosphate-dependent glycolysis, the ATP/ADP-dependent enzymes phosphofructokinase (PFK) and pyruvate kinase are replaced by the pyrophosphate-dependent PFK and pyruvate phosphate dikinase (PPDK), respectively. This variant of glycolysis is widespread among bacteria, but it also occurs in a few parasitic anaerobic eukaryotes such as Giardia and Entamoeba spp. We sequenced two genes for PPDK from the amitochondriate oxymonad Streblomastix strix and found evidence for PPDK in Trichomonas vaginalis and other parabasalia, where this enzyme was thought to be absent. The Streblomastix and Giardia genes may be related to one another, but those of Entamoeba and perhaps Trichomonas are distinct and more closely related to bacterial homologues. These findings suggest that pyrophosphate-dependent glycolysis is more widespread in eukaryotes than previously thought, enzymes from the pathway coexists with ATP-dependent more often than previously thought and may be spread by lateral transfer of genes for pyrophosphate-dependent enzymes from bacteria. Adaptation to anaerobic metabolism is a complex process (PPDK), respectively (for a comparison of these reactions, see involving changes to many proteins and pathways of critical reference 21). -

Characterization of a Eukaryotic Type Serine/Threonine Protein Kinase And
Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates Linda Nova´ kova´ 1, Lenka Saskova´ 1, Petra Pallova´ 1, Jirˇı´ Janecˇek1, Jana Novotna´ 1, Alesˇ Ulrych1, Jose Echenique2, Marie-Claude Trombe3 and Pavel Branny1 1 Cell and Molecular Microbiology Division, Institute of Microbiology, Czech Academy of Sciences, Prague, Czech Republic 2 Departamento de Bioquı´mica Clı´nica, Facultad de Ciencias Quı´micas, Universidad Nacional de Co´ rdoba, Medina Allende esq Haya de la Torre, Ciudad Universitaria, Co´ rdoba, Argentina 3 Centre Hospitalo-Universitaire de Rangueil, Universite´ Paul Sabatier, Toulouse, France Keywords Searching the genome sequence of Streptococcus pneumoniae revealed the phosphoglucosamine mutase; presence of a single Ser ⁄ Thr protein kinase gene stkP linked to protein phosphoproteome; protein phosphatase; phosphatase phpP. Biochemical studies performed with recombinant StkP serine ⁄ threonine protein kinase; suggest that this protein is a functional eukaryotic-type Ser ⁄ Thr protein Streptococcus pneumoniae kinase. In vitro kinase assays and Western blots of S. pneumoniae subcellu- Correspondence lar fractions revealed that StkP is a membrane protein. PhpP is a soluble P. Branny, Cell and Molecular Microbiology protein with manganese-dependent phosphatase activity in vitro against a Division, Institute of Microbiology, Czech synthetic substrate RRA(pT)VA. Mutations in the invariant aspartate resi- Academy of Sciences, Vı´denˇ ska´ 1083, dues implicated in the metal binding completely abolished PhpP activity. 142 20 Prague 4, Czech Republic Autophosphorylated form of StkP was shown to be a substrate for PhpP. Fax: +420 2 41722257 These results suggest that StkP and PhpP could operate as a functional Tel: +420 2 41062658 E-mail: [email protected] pair in vivo. -
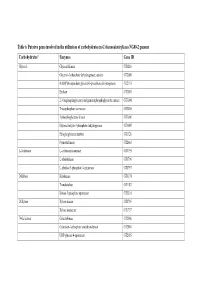
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

Evolving a New Efficient Mode of Fructose Utilization For
fbioe-09-669093 May 22, 2021 Time: 22:55 # 1 ORIGINAL RESEARCH published: 28 May 2021 doi: 10.3389/fbioe.2021.669093 Evolving a New Efficient Mode of Fructose Utilization for Improved Bioproduction in Corynebacterium glutamicum Irene Krahn1, Daniel Bonder2, Lucía Torregrosa-Barragán2, Dominik Stoppel1, 1 3 1 3,4 Edited by: Jens P. Krause , Natalie Rosenfeldt , Tobias M. Meiswinkel , Gerd M. Seibold , Pablo Ivan Nikel, Volker F. Wendisch1 and Steffen N. Lindner1,2* Novo Nordisk Foundation Center 1 2 for Biosustainability (DTU Biosustain), Chair of Genetics of Prokaryotes, Faculty of Biology and CeBiTec, Bielefeld University, Bielefeld, Germany, Systems 3 Denmark and Synthetic Metabolism, Max Planck Institute of Molecular Plant Physiology, Potsdam-Golm, Germany, Institute of Biochemistry, University of Cologne, Cologne, Germany, 4 Department of Biotechnology and Biomedicine, Technical Reviewed by: University of Denmark, Lyngby, Denmark Stephan Noack, Julich-Forschungszentrum, Helmholtz-Verband Deutscher Fructose utilization in Corynebacterium glutamicum starts with its uptake and Forschungszentren (HZ), Germany concomitant phosphorylation via the phosphotransferase system (PTS) to yield Fabien Létisse, UMR 5504 Laboratoire d’Ingénierie intracellular fructose 1-phosphate, which enters glycolysis upon ATP-dependent des Systèmes Biologiques et des phosphorylation to fructose 1,6-bisphosphate by 1-phosphofructokinase. This is known Procédés (LISBP), France to result in a significantly reduced oxidative pentose phosphate pathway (oxPPP) flux *Correspondence: ∼ ∼ Steffen N. Lindner on fructose ( 10%) compared to glucose ( 60%). Consequently, the biosynthesis of [email protected] NADPH demanding products, e.g., L-lysine, by C. glutamicum is largely decreased when fructose is the only carbon source. Previous works reported that fructose Specialty section: This article was submitted to is partially utilized via the glucose-specific PTS presumably generating fructose 6- Synthetic Biology, phosphate. -

Engineering Zymomonas Mobilis for the Production of Biofuels and Other Value-Added Products
© 2015 Kori Lynn Dunn ENGINEERING ZYMOMONAS MOBILIS FOR THE PRODUCTION OF BIOFUELS AND OTHER VALUE-ADDED PRODUCTS BY KORI LYNN DUNN DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Associate Professor Christopher V. Rao, Chair Associate Professor Yong-Su Jin Associate Professor Hyunjoon Kong Associate Professor Mary Kraft Abstract Zymomonas mobilis is a promising organism for lignocellulosic biofuel production as it can efficiently produce ethanol from simple sugars using unique metabolic pathways. Z. mobilis displays what is known as the “uncoupled growth” phenomenon, meaning cells will rapidly convert sugars to ethanol regardless of their energy requirements for growth. This makes Z. mobilis attractive not only for ethanol production, but for alternative product synthesis as well. One limitation of Z. mobilis for cellulosic ethanol production is that this organism cannot natively ferment the pentose sugars, like xylose and arabinose, which are present in lignocellulosic hydrolysates. While it has been engineered to do so, the fermentation rates of these sugars are still extremely low. In this work, we have investigated sugar transport as a possible bottleneck in the fermentation of xylose by Z. mobilis. We showed that transport limits xylose fermentations in this organism, but only when the starting sugar concentration is high. To discern additional bottlenecks in pentose fermentations by Z. mobilis, we then used adaptation and high-throughput sequencing to pinpoint genetic mutations responsible for improved growth phenotypes on these sugars. We found that the transport of both xylose and arabinose through the native sugar transporter, Glf, limits pentose fermentations in Z. -

Development and Control of Intestinal and Hepatic Fructokinase
INTESTINAL AND HEPATIC FRUCTOKINASE 765 expansion on intrarenal distribution of plasma flow in the dog. 42. The authors acknowledge the expert assistance of Mrs. Ann- Amer. J. Physiol., 223: 125 (1972). Christine Eklof and Mr. Gothe Carlebjork. 37. West, G. R., Smith, H. W., and Chasis, H.: Glomerular filtration 43. This research was supported by Grant no. B74-19X-3644-03Bfrom rate, effective renal blood flow and maximal tubular excretory the Swedish Medical Research Council and by a grant from the capacity in infancy. J. Pediat., 32: 10 (1948). Prenatal Research Funds of Expressen. 38. Laevosan Gesellschaft, Linz, Austria. 44. Requests for reprints should be addressed to: P. Herin, M.D., 39. 3M Company, St. Paul, Minn. Karolinski Institutet, Pediatriska Kliniken, St: Goran's Barn- 40. Sage Instruments, Inc., White Plains, N. Y. liniker, Box 12500, 112 81 Stockholm, Sweden. 41. Gilford Instrument Labs., Inc., Oberlin, Ohio. 45. Accepted for publication April 4, 1974. Copyright O 1974 International Pediatric Research Foundation, Inc. Printed in U.S.A. Pediat. Res. 8: 765-770 (1974) Developmental biochemistry intestine fetus liver fruct okinase Development and Control of Intestinal and Hepatic Fructokinase RICHARD J. GRAND,(^') MARIA I. SCHAY, AND STEPHANIE JAKSINA Department of Pediatrics, Harvard Medical School, and the Department of Medicine (Division of Clinical Nutrition), the Children's Hospital Medical Center, Boston, Massachusetts, USA Extract experimental animals (1, 2, 20, 26) and in man (I 5), although the mechanism of this regulation has not yet been elucidated. The patterns of development of intestinal and hepatic In the rat, adrenalectomy prevents the rise in the activity of fructokinase have been studied in the rat, in the rabbit, and in the hepatic enzyme expected after fructose feeding; hydro- man, and information regarding the mechanism of control of cortisone administered to the intact animal greatly enhances this enzyme in mature organs has been obtained. -

Of Candida Bombicola
Aerodynamically, the bumble bee shouldn't be able to fly, but the bumble bee doesn't know it so it goes on flying anyway. Mary Kay Ash Jury: Prof. Dr. ir. Norbert DE KIMPE Prof. Dr. ir. Nico BOON Lic. Dirk DEVELTER Prof. Dr. ir. Monica HÖFTE Prof. Dr. Andreas SCHMID Prof. Dr. Els VAN DAMME Prof. Dr. ir. Wim SOETAERT Prof. Dr. ir. Erick VANDAMME Promotors: Prof . Dr. ir. Erick VANDAMME Prof. Dr. ir. Wim SOETAERT Laboratory of Industrial Microbiology and Biocatalysis Department of Biochemical and Microbial Technology Ghent University Dean: Prof. Dr. ir. Herman VAN LANGENHOVE Rector: Prof. Dr. Paul VAN CAUWENBERGE Ir. Inge Van Bogaert was supported by Ecover Belgium NV (Malle, Belgium) and a fellowship of the Bijzonder Onderzoekfonds of Ghent University (BOF). The research was conducted at the Laboratory of Industrial Microbiology and Biocatalysis, Department of Biochemical and Microbial Technology, Ghent University. ir. Inge Van Bogaert MICROBIAL SYNTHESIS OF SOPHOROLIPIDS BY THE YEAST CANDIDA BOMBICOLA Thesis submitted in fulfillment of the requirements for the degree of Doctor (PhD) in Applied Biological Sciences Titel van het doctoraatsproefschrift in het Nederlands: Microbiële synthese van sopohorolipiden door de gist Candida bombicola Cover illustration: Cadzand on a stormy day by Inge Van Bogaert Refer to this thesis: Van Bogaert INA (2008) Microbial synthesis of sophorolipids by the yeast Candida bombicola. PhD-thesis, Faculty of Bioscience Engineering, Ghent University, Ghent, Belgium, 239 p. ISBN-number: ISBN 978-90-5989-243-9 The author and the promotor give the authorisation to consult and to copy parts of this work for personal use only. -

Characterization of Ribose-5-Phosphate Isomerase B from Newly Isolated Strain Ochrobactrum Sp
J. Microbiol. Biotechnol. (2018), 28(7), 1122–1132 https://doi.org/10.4014/jmb.1802.02021 Research Article Review jmb Characterization of Ribose-5-Phosphate Isomerase B from Newly Isolated Strain Ochrobactrum sp. CSL1 Producing L-Rhamnulose from L-Rhamnose Min Shen1†, Xin Ju1†, Xinqi Xu2, Xuemei Yao1, Liangzhi Li1*, Jiajia Chen1, Cuiying Hu1, Jiaolong Fu1, and Lishi Yan1 1School of Chemistry, Biology, and Material Engineering, Suzhou University of Science and Technology, Suzhou 215009, P.R. China 2Fujian Key Laboratory of Marine Enzyme Engineering, Fuzhou University, Fujian 350116, P.R. China Received: February 14, 2018 Revised: April 12, 2018 In this study, we attempted to find new and efficient microbial enzymes for producing rare Accepted: April 13, 2018 sugars. A ribose-5-phosphate isomerase B (OsRpiB) was cloned, overexpressed, and First published online preliminarily purified successfully from a newly screened Ochrobactrum sp. CSL1, which could May 8, 2018 catalyze the isomerization reaction of rare sugars. A study of its substrate specificity showed *Corresponding author that the cloned isomerase (OsRpiB) could effectively catalyze the conversion of L-rhamnose to Phone: +86-512-68056493; L-rhamnulose, which was unconventional for RpiB. The optimal reaction conditions (50oC, Fax: +86-512-68418431; 2+ E-mail: [email protected] pH 8.0, and 1 mM Ca ) were obtained to maximize the potential of OsRpiB in preparing L-rhamnulose. The catalytic properties of OsRpiB, including Km, kcat, and catalytic efficiency † These authors contributed (k /K ), were determined as 43.47 mM, 129.4 sec-1, and 2.98 mM/sec. The highest conversion equally to this work. -

Regulation of Fructose-6-Phosphate 2-Kinase By
Proc. Natt Acad. Sci. USA Vol. 79, pp. 325-329, January 1982 Biochemistry Regulation of fructose-6-phosphate 2-kinase by phosphorylation and dephosphorylation: Possible mechanism for coordinated control of glycolysis and glycogenolysis (phosphofructokinase) EISUKE FURUYA*, MOTOKO YOKOYAMA, AND KOSAKU UYEDAt Pre-Clinical Science Unit of the Veterans Administration Medical Center, 4500 South Lancaster Road, Dallas, Texas 75216; and Biochemistry Department of the University ofTexas Health Science Center, 5323 Harry Hines Boulevard, Dallas, Texas 75235 Communicated by Jesse C. Rabinowitz, September 28, 1981 ABSTRACT The kinetic properties and the control mecha- Fructose 6-phosphate + ATP nism of fructose-6-phosphate 2-kinase (ATP: D-fructose-6-phos- -3 Fructose + ADP. [1] phate 2-phosphotransferase) were investigated. The molecular 2,6-bisphosphate weight of the enzyme is -100,000 as determined by gel filtration. The plot of initial velocity versus ATP concentration is hyperbolic We have shown that the administration of extremely low con- with a K. of 1.2 mM. However, the plot of enzyme activity as a centrations of glucagon (0.1 fM) or high concentrations of epi- function of fructose 6-phosphate is sigmoidal. The apparent K0.5 nephrine (10 ,uM) to hepatocytes results in inactivation offruc- for fructose 6-phosphate is 20 ,IM. Fructose-6-phosphate 2-kinase tose-6-phosphate 2-kinase and concomitant decrease in the is inactivated by -the catalytic subunit of cyclic AMP-dependent fructose 2,6-bisphosphate level (12). These results, as well as protein kinase, and the inactivation is closely correlated with phos- more recent data using Ca2+ and the Ca2+ ionophore A23187 phorylation.