Two Structures of the Catalytic Domain of Phosphorylase Kinase
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

(12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk Et Al
US 2014O155567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk et al. (43) Pub. Date: Jun. 5, 2014 (54) MICROORGANISMS AND METHODS FOR (60) Provisional application No. 61/331,812, filed on May THE BIOSYNTHESIS OF BUTADENE 5, 2010. (71) Applicant: Genomatica, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Mark J. Burk, San Diego, CA (US); (51) Int. Cl. Anthony P. Burgard, Bellefonte, PA CI2P 5/02 (2006.01) (US); Jun Sun, San Diego, CA (US); CSF 36/06 (2006.01) Robin E. Osterhout, San Diego, CA CD7C II/6 (2006.01) (US); Priti Pharkya, San Diego, CA (52) U.S. Cl. (US) CPC ................. CI2P5/026 (2013.01); C07C II/I6 (2013.01); C08F 136/06 (2013.01) (73) Assignee: Genomatica, Inc., San Diego, CA (US) USPC ... 526/335; 435/252.3:435/167; 435/254.2: (21) Appl. No.: 14/059,131 435/254.11: 435/252.33: 435/254.21:585/16 (22) Filed: Oct. 21, 2013 (57) ABSTRACT O O The invention provides non-naturally occurring microbial Related U.S. Application Data organisms having a butadiene pathway. The invention addi (63) Continuation of application No. 13/101,046, filed on tionally provides methods of using Such organisms to produce May 4, 2011, now Pat. No. 8,580,543. butadiene. Patent Application Publication Jun. 5, 2014 Sheet 1 of 4 US 2014/O155567 A1 ?ueudos!SMS |?un61– Patent Application Publication Jun. 5, 2014 Sheet 2 of 4 US 2014/O155567 A1 VOJ OO O Z?un61– Patent Application Publication US 2014/O155567 A1 {}}} Hººso Patent Application Publication Jun. -

Glycogen Synthase Kinase-3 Is Activated in Neuronal Cells by G 12
The Journal of Neuroscience, August 15, 2002, 22(16):6863–6875 Glycogen Synthase Kinase-3 Is Activated in Neuronal Cells by G␣ ␣ 12 and G 13 by Rho-Independent and Rho-Dependent Mechanisms C. Laura Sayas, Jesu´ s Avila, and Francisco Wandosell Centro de Biologı´a Molecular “Severo Ochoa”, Consejo Superior de Investigaciones Cientı´ficas, Universidad Auto´ noma de Madrid, Cantoblanco, Madrid 28049, Spain ␣ ␣ ␣ ␣ Glycogen synthase kinase-3 (GSK-3) was generally considered tively active G 12 (G 12QL) and G 13 (G 13QL) in Neuro2a cells a constitutively active enzyme, only regulated by inhibition. induces upregulation of GSK-3 activity. Furthermore, overex- Here we describe that GSK-3 is activated by lysophosphatidic pression of constitutively active RhoA (RhoAV14) also activates ␣ acid (LPA) during neurite retraction in rat cerebellar granule GSK-3 However, the activation of GSK-3 by G 13 is blocked by neurons. GSK-3 activation correlates with an increase in GSK-3 coexpression with C3 transferase, whereas C3 does not block ␣ tyrosine phosphorylation. In addition, LPA induces a GSK-3- GSK-3 activation by G 12. Thus, we demonstrate that GSK-3 is ␣ ␣ mediated hyperphosphorylation of the microtubule-associated activated by both G 12 and G 13 in neuronal cells. However, ␣ protein tau. Inhibition of GSK-3 by lithium partially blocks neu- GSK-3 activation by G 13 is Rho-mediated, whereas GSK-3 ␣ rite retraction, indicating that GSK-3 activation is important but activation by G 12 is Rho-independent. The results presented not essential for the neurite retraction progress. GSK-3 activa- here imply the existence of a previously unknown mechanism of ␣ tion by LPA in cerebellar granule neurons is neither downstream GSK-3 activation by G 12/13 subunits. -

Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides
catalysts Review α-Glucan Phosphorylase-Catalyzed Enzymatic Reactions Using Analog Substrates to Synthesize Non-Natural Oligo- and Polysaccharides Jun-ichi Kadokawa Department of Chemistry, Biotechnology, and Chemical Engineering, Graduate School of Science and Engineering, Kagoshima University, 1-21-40 Korimoto, Kagoshima 860-0065, Japan; [email protected]; Tel.: +81-99-285-7743 Received: 9 October 2018; Accepted: 16 October 2018; Published: 19 October 2018 Abstract: As natural oligo- and polysaccharides are important biomass resources and exhibit vital biological functions, non-natural oligo- and polysaccharides with a well-defined structure can be expected to act as new functional materials with specific natures and properties. α-Glucan phosphorylase (GP) is one of the enzymes that have been used as catalysts for practical synthesis of oligo- and polysaccharides. By means of weak specificity for the recognition of substrates by GP, non-natural oligo- and polysaccharides has precisely been synthesized. GP-catalyzed enzymatic glycosylations using several analog substrates as glycosyl donors have been carried out to produce oligosaccharides having different monosaccharide residues at the non-reducing end. Glycogen, a highly branched natural polysaccharide, has been used as the polymeric glycosyl acceptor and primer for the GP-catalyzed glycosylation and polymerization to obtain glycogen-based non-natural polysaccharide materials. Under the conditions of removal of inorganic phosphate, thermostable GP-catalyzed enzymatic polymerization of analog monomers occurred to give amylose analog polysaccharides. Keywords: analog substrate; α-glucan phosphorylase; non-natural oligo- and polysaccharides 1. Introduction Oligo- and polysaccharides are widely distributed in nature and enact specific important biological functions in accordance with their chemical structures [1]. -

Defective Galactose Oxidation in a Patient with Glycogen Storage Disease and Fanconi Syndrome
Pediatr. Res. 17: 157-161 (1983) Defective Galactose Oxidation in a Patient with Glycogen Storage Disease and Fanconi Syndrome M. BRIVET,"" N. MOATTI, A. CORRIAT, A. LEMONNIER, AND M. ODIEVRE Laboratoire Central de Biochimie du Centre Hospitalier de Bichre, 94270 Kremlin-Bicetre, France [M. B., A. C.]; Faculte des Sciences Pharmaceutiques et Biologiques de I'Universite Paris-Sud, 92290 Chatenay-Malabry, France [N. M., A. L.]; and Faculte de Midecine de I'Universiti Paris-Sud et Unite de Recherches d'Hepatologie Infantile, INSERM U 56, 94270 Kremlin-Bicetre. France [M. 0.1 Summary The patient's diet was supplemented with 25-OH-cholecalci- ferol, phosphorus, calcium, and bicarbonate. With this treatment, Carbohydrate metabolism was studied in a child with atypical the serum phosphate concentration increased, but remained be- glycogen storage disease and Fanconi syndrome. Massive gluco- tween 0.8 and 1.0 mmole/liter, whereas the plasma carbon dioxide suria, partial resistance to glucagon and abnormal responses to level returned to normal (18-22 mmole/liter). Rickets was only carbohydrate loads, mainly in the form of major impairment of partially controlled. galactose utilization were found, as reported in previous cases. Increased blood lactate to pyruvate ratios, observed in a few cases of idiopathic Fanconi syndrome, were not present. [l-14ClGalac- METHODS tose oxidation was normal in erythrocytes, but reduced in fresh All studies of the patient and of the subjects who served as minced liver tissue, despite normal activities of hepatic galactoki- controls were undertaken after obtaining parental or personal nase, uridyltransferase, and UDP-glucose 4epirnerase in hornog- consent. enates of frozen liver. -

Pyruvate-Phosphate Dikinase of Oxymonads and Parabasalia and the Evolution of Pyrophosphate-Dependent Glycolysis in Anaerobic Eukaryotes† Claudio H
EUKARYOTIC CELL, Jan. 2006, p. 148–154 Vol. 5, No. 1 1535-9778/06/$08.00ϩ0 doi:10.1128/EC.5.1.148–154.2006 Copyright © 2006, American Society for Microbiology. All Rights Reserved. Pyruvate-Phosphate Dikinase of Oxymonads and Parabasalia and the Evolution of Pyrophosphate-Dependent Glycolysis in Anaerobic Eukaryotes† Claudio H. Slamovits and Patrick J. Keeling* Canadian Institute for Advanced Research, Botany Department, University of British Columbia, 3529-6270 University Boulevard, Vancouver, British Columbia V6T 1Z4, Canada Received 29 September 2005/Accepted 8 November 2005 In pyrophosphate-dependent glycolysis, the ATP/ADP-dependent enzymes phosphofructokinase (PFK) and pyruvate kinase are replaced by the pyrophosphate-dependent PFK and pyruvate phosphate dikinase (PPDK), respectively. This variant of glycolysis is widespread among bacteria, but it also occurs in a few parasitic anaerobic eukaryotes such as Giardia and Entamoeba spp. We sequenced two genes for PPDK from the amitochondriate oxymonad Streblomastix strix and found evidence for PPDK in Trichomonas vaginalis and other parabasalia, where this enzyme was thought to be absent. The Streblomastix and Giardia genes may be related to one another, but those of Entamoeba and perhaps Trichomonas are distinct and more closely related to bacterial homologues. These findings suggest that pyrophosphate-dependent glycolysis is more widespread in eukaryotes than previously thought, enzymes from the pathway coexists with ATP-dependent more often than previously thought and may be spread by lateral transfer of genes for pyrophosphate-dependent enzymes from bacteria. Adaptation to anaerobic metabolism is a complex process (PPDK), respectively (for a comparison of these reactions, see involving changes to many proteins and pathways of critical reference 21). -

Characterization of a Eukaryotic Type Serine/Threonine Protein Kinase And
Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates Linda Nova´ kova´ 1, Lenka Saskova´ 1, Petra Pallova´ 1, Jirˇı´ Janecˇek1, Jana Novotna´ 1, Alesˇ Ulrych1, Jose Echenique2, Marie-Claude Trombe3 and Pavel Branny1 1 Cell and Molecular Microbiology Division, Institute of Microbiology, Czech Academy of Sciences, Prague, Czech Republic 2 Departamento de Bioquı´mica Clı´nica, Facultad de Ciencias Quı´micas, Universidad Nacional de Co´ rdoba, Medina Allende esq Haya de la Torre, Ciudad Universitaria, Co´ rdoba, Argentina 3 Centre Hospitalo-Universitaire de Rangueil, Universite´ Paul Sabatier, Toulouse, France Keywords Searching the genome sequence of Streptococcus pneumoniae revealed the phosphoglucosamine mutase; presence of a single Ser ⁄ Thr protein kinase gene stkP linked to protein phosphoproteome; protein phosphatase; phosphatase phpP. Biochemical studies performed with recombinant StkP serine ⁄ threonine protein kinase; suggest that this protein is a functional eukaryotic-type Ser ⁄ Thr protein Streptococcus pneumoniae kinase. In vitro kinase assays and Western blots of S. pneumoniae subcellu- Correspondence lar fractions revealed that StkP is a membrane protein. PhpP is a soluble P. Branny, Cell and Molecular Microbiology protein with manganese-dependent phosphatase activity in vitro against a Division, Institute of Microbiology, Czech synthetic substrate RRA(pT)VA. Mutations in the invariant aspartate resi- Academy of Sciences, Vı´denˇ ska´ 1083, dues implicated in the metal binding completely abolished PhpP activity. 142 20 Prague 4, Czech Republic Autophosphorylated form of StkP was shown to be a substrate for PhpP. Fax: +420 2 41722257 These results suggest that StkP and PhpP could operate as a functional Tel: +420 2 41062658 E-mail: [email protected] pair in vivo. -
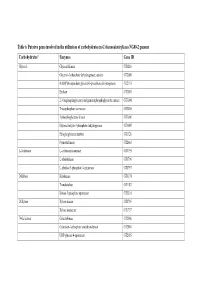
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

Evolving a New Efficient Mode of Fructose Utilization For
fbioe-09-669093 May 22, 2021 Time: 22:55 # 1 ORIGINAL RESEARCH published: 28 May 2021 doi: 10.3389/fbioe.2021.669093 Evolving a New Efficient Mode of Fructose Utilization for Improved Bioproduction in Corynebacterium glutamicum Irene Krahn1, Daniel Bonder2, Lucía Torregrosa-Barragán2, Dominik Stoppel1, 1 3 1 3,4 Edited by: Jens P. Krause , Natalie Rosenfeldt , Tobias M. Meiswinkel , Gerd M. Seibold , Pablo Ivan Nikel, Volker F. Wendisch1 and Steffen N. Lindner1,2* Novo Nordisk Foundation Center 1 2 for Biosustainability (DTU Biosustain), Chair of Genetics of Prokaryotes, Faculty of Biology and CeBiTec, Bielefeld University, Bielefeld, Germany, Systems 3 Denmark and Synthetic Metabolism, Max Planck Institute of Molecular Plant Physiology, Potsdam-Golm, Germany, Institute of Biochemistry, University of Cologne, Cologne, Germany, 4 Department of Biotechnology and Biomedicine, Technical Reviewed by: University of Denmark, Lyngby, Denmark Stephan Noack, Julich-Forschungszentrum, Helmholtz-Verband Deutscher Fructose utilization in Corynebacterium glutamicum starts with its uptake and Forschungszentren (HZ), Germany concomitant phosphorylation via the phosphotransferase system (PTS) to yield Fabien Létisse, UMR 5504 Laboratoire d’Ingénierie intracellular fructose 1-phosphate, which enters glycolysis upon ATP-dependent des Systèmes Biologiques et des phosphorylation to fructose 1,6-bisphosphate by 1-phosphofructokinase. This is known Procédés (LISBP), France to result in a significantly reduced oxidative pentose phosphate pathway (oxPPP) flux *Correspondence: ∼ ∼ Steffen N. Lindner on fructose ( 10%) compared to glucose ( 60%). Consequently, the biosynthesis of [email protected] NADPH demanding products, e.g., L-lysine, by C. glutamicum is largely decreased when fructose is the only carbon source. Previous works reported that fructose Specialty section: This article was submitted to is partially utilized via the glucose-specific PTS presumably generating fructose 6- Synthetic Biology, phosphate. -

Engineering Zymomonas Mobilis for the Production of Biofuels and Other Value-Added Products
© 2015 Kori Lynn Dunn ENGINEERING ZYMOMONAS MOBILIS FOR THE PRODUCTION OF BIOFUELS AND OTHER VALUE-ADDED PRODUCTS BY KORI LYNN DUNN DISSERTATION Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering in the Graduate College of the University of Illinois at Urbana-Champaign, 2015 Urbana, Illinois Doctoral Committee: Associate Professor Christopher V. Rao, Chair Associate Professor Yong-Su Jin Associate Professor Hyunjoon Kong Associate Professor Mary Kraft Abstract Zymomonas mobilis is a promising organism for lignocellulosic biofuel production as it can efficiently produce ethanol from simple sugars using unique metabolic pathways. Z. mobilis displays what is known as the “uncoupled growth” phenomenon, meaning cells will rapidly convert sugars to ethanol regardless of their energy requirements for growth. This makes Z. mobilis attractive not only for ethanol production, but for alternative product synthesis as well. One limitation of Z. mobilis for cellulosic ethanol production is that this organism cannot natively ferment the pentose sugars, like xylose and arabinose, which are present in lignocellulosic hydrolysates. While it has been engineered to do so, the fermentation rates of these sugars are still extremely low. In this work, we have investigated sugar transport as a possible bottleneck in the fermentation of xylose by Z. mobilis. We showed that transport limits xylose fermentations in this organism, but only when the starting sugar concentration is high. To discern additional bottlenecks in pentose fermentations by Z. mobilis, we then used adaptation and high-throughput sequencing to pinpoint genetic mutations responsible for improved growth phenotypes on these sugars. We found that the transport of both xylose and arabinose through the native sugar transporter, Glf, limits pentose fermentations in Z. -

Chem331 Glycogen Metabolism
Glycogen metabolism Glycogen review - 1,4 and 1,6 α-glycosidic links ~ every 10 sugars are branched - open helix with many non-reducing ends. Effective storage of glucose Glucose storage Liver glycogen 4.0% 72 g Muscle glycogen 0.7% 245 g Blood Glucose 0.1% 10 g Large amount of water associated with glycogen - 0.5% of total weight Glycogen stored in granules in cytosol w/proteins for synthesis, degradation and control There are very different means of control of glycogen metabolism between liver and muscle Glycogen biosynthetic and degradative cycle Two different pathways - which do not share enzymes like glycolysis and gluconeogenesis glucose -> glycogen glycogenesis - biosynthetic glycogen -> glucose 1-P glycogenolysis - breakdown Evidence for two paths - Patients lacking phosphorylase can still synthesize glycogen - hormonal regulation of both directions Glycogenolysis (glycogen breakdown)- Glycogen Phosphorylase glycogen (n) + Pi -> glucose 1-p + glycogen (n-1) • Enzyme binds and cleaves glycogen into monomers at the end of the polymer (reducing ends of glycogen) • Dimmer interacting at the N-terminus. • rate limiting - controlled step in glycogen breakdown • glycogen phosphorylase - cleavage of 1,4 α glycosidic bond by Pi NOT H2O • Energy of phosphorolysis vs. hydrolysis -low standard state free energy change -transfer potential -driven by Pi concentration -Hydrolysis would require additional step s/ cost of ATP - Think of the difference between adding a phosphate group with hydrolysis • phosphorylation locks glucose in cell (imp. for muscle) • Phosphorylase binds glycogen at storage site and the catalytic site is 4 to 5 glucose residues away from the catalytic site. • Phosphorylase removes 1 residue at a time from glycogen until 4 glucose residues away on either side of 1,6 branch point – stericaly hindered by glycogen storage site • Cleaves without releasing at storage site • general acid/base catalysts • Inorganic phosphate attacks the terminal glucose residue passing through an oxonium ion intermediate. -

The Origins of Protein Phosphorylation
historical perspective The origins of protein phosphorylation Philip Cohen The reversible phosphorylation of proteins is central to the regulation of most aspects of cell func- tion but, even after the first protein kinase was identified, the general significance of this discovery was slow to be appreciated. Here I review the discovery of protein phosphorylation and give a per- sonal view of the key findings that have helped to shape the field as we know it today. he days when protein phosphorylation was an abstruse backwater, best talked Tabout between consenting adults in private, are over. My colleagues no longer cringe on hearing that “phosphorylase kinase phosphorylates phosphorylase” and their eyes no longer glaze over when a “”kinase kinase kinase” is mentioned. This is because protein phosphorylation has gradu- ally become an integral part of all the sys- tems they are studying themselves. Indeed it would be difficult to find anyone today who would disagree with the statement that “the reversible phosphorylation of proteins regu- lates nearly every aspect of cell life”. Phosphorylation and dephosphorylation, catalysed by protein kinases and protein phosphatases, can modify the function of a protein in almost every conceivable way; for Carl and Gerty Cori, the 1947 Nobel Laureates. Picture: Science Photo Library. example by increasing or decreasing its bio- logical activity, by stabilizing it or marking it for destruction, by facilitating or inhibiting movement between subcellular compart- so long before its general significance liver enzyme that catalysed the phosphory- ments, or by initiating or disrupting pro- was appreciated? lation of casein3. Soon after, Fischer and tein–protein interactions.