Engineering Zymomonas Mobilis for the Production of Biofuels and Other Value-Added Products
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Xylose Fermentation to Ethanol by Schizosaccharomyces Pombe Clones with Xylose Isomerase Gene." Biotechnology Letters (8:4); Pp
NREL!TP-421-4944 • UC Category: 246 • DE93000067 l I Xylose Fermenta to Ethanol: A R ew '.) i I, -- , ) )I' J. D. McMillan I ' J.( .!i �/ .6' ....� .T u�.•ls:l ., �-- • National Renewable Energy Laboratory II 'J 1617 Cole Boulevard Golden, Colorado 80401-3393 A Division of Midwest Research Institute Operated for the U.S. Department of Energy under Contract No. DE-AC02-83CH10093 Prepared under task no. BF223732 January 1993 NOTICE This report was prepared as an account of work sponsored by an agency of the United States government. Neither the United States government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, com pleteness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily con stitute or imply its endorsement, recommendation, or favoring by the United States government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States government or any agency thereof. Printed in the United States of America Available from: National Technical Information Service U.S. Department of Commerce 5285 Port Royal Road Springfield, VA22161 Price: Microfiche A01 Printed Copy A03 Codes are used for pricing all publications. The code is determined by the number of pages in the publication. Information pertaining to the pricing codes can be found in the current issue of the following publications which are generally available in most libraries: Energy Research Abstracts (ERA); Govern ment Reports Announcements and Index ( GRA and I); Scientific and Technical Abstract Reports(STAR); and publication NTIS-PR-360 available from NTIS at the above address. -

(12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk Et Al
US 2014O155567A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0155567 A1 Burk et al. (43) Pub. Date: Jun. 5, 2014 (54) MICROORGANISMS AND METHODS FOR (60) Provisional application No. 61/331,812, filed on May THE BIOSYNTHESIS OF BUTADENE 5, 2010. (71) Applicant: Genomatica, Inc., San Diego, CA (US) Publication Classification (72) Inventors: Mark J. Burk, San Diego, CA (US); (51) Int. Cl. Anthony P. Burgard, Bellefonte, PA CI2P 5/02 (2006.01) (US); Jun Sun, San Diego, CA (US); CSF 36/06 (2006.01) Robin E. Osterhout, San Diego, CA CD7C II/6 (2006.01) (US); Priti Pharkya, San Diego, CA (52) U.S. Cl. (US) CPC ................. CI2P5/026 (2013.01); C07C II/I6 (2013.01); C08F 136/06 (2013.01) (73) Assignee: Genomatica, Inc., San Diego, CA (US) USPC ... 526/335; 435/252.3:435/167; 435/254.2: (21) Appl. No.: 14/059,131 435/254.11: 435/252.33: 435/254.21:585/16 (22) Filed: Oct. 21, 2013 (57) ABSTRACT O O The invention provides non-naturally occurring microbial Related U.S. Application Data organisms having a butadiene pathway. The invention addi (63) Continuation of application No. 13/101,046, filed on tionally provides methods of using Such organisms to produce May 4, 2011, now Pat. No. 8,580,543. butadiene. Patent Application Publication Jun. 5, 2014 Sheet 1 of 4 US 2014/O155567 A1 ?ueudos!SMS |?un61– Patent Application Publication Jun. 5, 2014 Sheet 2 of 4 US 2014/O155567 A1 VOJ OO O Z?un61– Patent Application Publication US 2014/O155567 A1 {}}} Hººso Patent Application Publication Jun. -

Pyruvate-Phosphate Dikinase of Oxymonads and Parabasalia and the Evolution of Pyrophosphate-Dependent Glycolysis in Anaerobic Eukaryotes† Claudio H
EUKARYOTIC CELL, Jan. 2006, p. 148–154 Vol. 5, No. 1 1535-9778/06/$08.00ϩ0 doi:10.1128/EC.5.1.148–154.2006 Copyright © 2006, American Society for Microbiology. All Rights Reserved. Pyruvate-Phosphate Dikinase of Oxymonads and Parabasalia and the Evolution of Pyrophosphate-Dependent Glycolysis in Anaerobic Eukaryotes† Claudio H. Slamovits and Patrick J. Keeling* Canadian Institute for Advanced Research, Botany Department, University of British Columbia, 3529-6270 University Boulevard, Vancouver, British Columbia V6T 1Z4, Canada Received 29 September 2005/Accepted 8 November 2005 In pyrophosphate-dependent glycolysis, the ATP/ADP-dependent enzymes phosphofructokinase (PFK) and pyruvate kinase are replaced by the pyrophosphate-dependent PFK and pyruvate phosphate dikinase (PPDK), respectively. This variant of glycolysis is widespread among bacteria, but it also occurs in a few parasitic anaerobic eukaryotes such as Giardia and Entamoeba spp. We sequenced two genes for PPDK from the amitochondriate oxymonad Streblomastix strix and found evidence for PPDK in Trichomonas vaginalis and other parabasalia, where this enzyme was thought to be absent. The Streblomastix and Giardia genes may be related to one another, but those of Entamoeba and perhaps Trichomonas are distinct and more closely related to bacterial homologues. These findings suggest that pyrophosphate-dependent glycolysis is more widespread in eukaryotes than previously thought, enzymes from the pathway coexists with ATP-dependent more often than previously thought and may be spread by lateral transfer of genes for pyrophosphate-dependent enzymes from bacteria. Adaptation to anaerobic metabolism is a complex process (PPDK), respectively (for a comparison of these reactions, see involving changes to many proteins and pathways of critical reference 21). -

Characterization of a Eukaryotic Type Serine/Threonine Protein Kinase And
Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates Linda Nova´ kova´ 1, Lenka Saskova´ 1, Petra Pallova´ 1, Jirˇı´ Janecˇek1, Jana Novotna´ 1, Alesˇ Ulrych1, Jose Echenique2, Marie-Claude Trombe3 and Pavel Branny1 1 Cell and Molecular Microbiology Division, Institute of Microbiology, Czech Academy of Sciences, Prague, Czech Republic 2 Departamento de Bioquı´mica Clı´nica, Facultad de Ciencias Quı´micas, Universidad Nacional de Co´ rdoba, Medina Allende esq Haya de la Torre, Ciudad Universitaria, Co´ rdoba, Argentina 3 Centre Hospitalo-Universitaire de Rangueil, Universite´ Paul Sabatier, Toulouse, France Keywords Searching the genome sequence of Streptococcus pneumoniae revealed the phosphoglucosamine mutase; presence of a single Ser ⁄ Thr protein kinase gene stkP linked to protein phosphoproteome; protein phosphatase; phosphatase phpP. Biochemical studies performed with recombinant StkP serine ⁄ threonine protein kinase; suggest that this protein is a functional eukaryotic-type Ser ⁄ Thr protein Streptococcus pneumoniae kinase. In vitro kinase assays and Western blots of S. pneumoniae subcellu- Correspondence lar fractions revealed that StkP is a membrane protein. PhpP is a soluble P. Branny, Cell and Molecular Microbiology protein with manganese-dependent phosphatase activity in vitro against a Division, Institute of Microbiology, Czech synthetic substrate RRA(pT)VA. Mutations in the invariant aspartate resi- Academy of Sciences, Vı´denˇ ska´ 1083, dues implicated in the metal binding completely abolished PhpP activity. 142 20 Prague 4, Czech Republic Autophosphorylated form of StkP was shown to be a substrate for PhpP. Fax: +420 2 41722257 These results suggest that StkP and PhpP could operate as a functional Tel: +420 2 41062658 E-mail: [email protected] pair in vivo. -
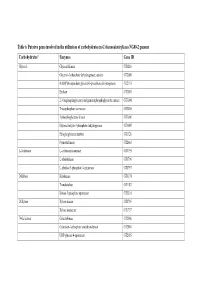
Table 6. Putative Genes Involved in the Utilization of Carbohydrates in G
Table 6. Putative genes involved in the utilization of carbohydrates in G. thermodenitrificans NG80-2 genome Carbohydrates* Enzymes Gene ID Glycerol Glycerol Kinase GT1216 Glycerol-3-phosphate dehydrogenase, aerobic GT2089 NAD(P)H-dependent glycerol-3-phosphate dehydrogenase GT2153 Enolase GT3003 2,3-bisphosphoglycerate-independentphosphoglycerate mutase GT3004 Triosephosphate isomerase GT3005 3-phosphoglycerate kinase GT3006 Glyceraldehyde-3-phosphate dehydrogenase GT3007 Phosphoglycerate mutase GT1326 Pyruvate kinase GT2663 L-Arabinose L-arabinose isomerase GT1795 L-ribulokinase GT1796 L-ribulose 5-phosphate 4-epimerase GT1797 D-Ribose Ribokinase GT3174 Transketolase GT1187 Ribose 5-phosphate epimerase GT3316 D-Xylose Xylose kinase GT1756 Xylose isomerase GT1757 D-Galactose Galactokinase GT2086 Galactose-1-phosphate uridyltransferase GT2084 UDP-glucose 4-epimerase GT2085 Carbohydrates* Enzymes Gene ID D-Fructose 1-phosphofructokinase GT1727 Fructose-1,6-bisphosphate aldolase GT1805 Fructose-1,6-bisphosphate aldolase type II GT3331 Triosephosphate isomerase GT3005 D-Mannose Mannnose-6 phospate isomelase GT3398 6-phospho-1-fructokinase GT2664 D-Mannitol Mannitol-1-phosphate dehydrogenase GT1844 N-Acetylglucosamine N-acetylglucosamine-6-phosphate deacetylase GT2205 N-acetylglucosamine-6-phosphate isomerase GT2204 D-Maltose Alpha-1,4-glucosidase GT0528, GT1643 Sucrose Sucrose phosphorylase GT3215 D-Trehalose Alpha-glucosidase GT1643 Glucose kinase GT2381 Inositol Myo-inositol catabolism protein iolC;5-dehydro-2- GT1807 deoxygluconokinase -

Zymomonas Mobilis in Bread Dough: Characterization of Dough Leavening Performance in Presence of Sucrose
foods Article Zymomonas mobilis in Bread Dough: Characterization of Dough Leavening Performance in Presence of Sucrose Alida Musatti * , Carola Cappa * , Chiara Mapelli, Cristina Alamprese and Manuela Rollini Dipartimento di Scienze per gli Alimenti, la Nutrizione, l’Ambiente, Università degli Studi di Milano, Via G. Celoria, 2-20133 Milano, Italy; [email protected] (C.M.); [email protected] (C.A.); [email protected] (M.R.) * Correspondence: [email protected] (A.M.); [email protected] (C.C.); Tel.: +39-02-5031-9150 (A.M.); +39-02-5031-9179 (C.C.) Received: 21 December 2019; Accepted: 11 January 2020; Published: 15 January 2020 Abstract: Zymomonas mobilis, because of its fermentative metabolism, has potential food applications in the development of leavened baked goods consumable by people with adverse responses to Saccharomyces cerevisiae. Since Z. mobilis is not able to utilize maltose present in flour, the effect of sucrose addition (2.5 g/100 g flour) on bread dough leavening properties was studied. For comparison purposes, leavening performances of S. cerevisiae with and without sucrose were also investigated. Doughs leavened by Z. mobilis without sucrose addition showed the lowest height development (14.95 0.21 mm) and CO production (855 136 mL). When sucrose was added, fermentative ± 2 ± performances of Z. mobilis significantly (p < 0.05) improved (+80% and +85% of gas production and retention, respectively), with a dough maximum height 2.6 times higher, results indicating that Z. mobilis with sucrose can be leavened in shorter time with respect to the sample without addition. S. cerevisiae did not benefit the sucrose addition in terms of CO2 production and retention, even if lag leavening time was significantly (p < 0.05) shorter (about the half) and time of porosity appearance significantly (p < 0.05) longer (about 26%) with respect to S. -

Evolving a New Efficient Mode of Fructose Utilization For
fbioe-09-669093 May 22, 2021 Time: 22:55 # 1 ORIGINAL RESEARCH published: 28 May 2021 doi: 10.3389/fbioe.2021.669093 Evolving a New Efficient Mode of Fructose Utilization for Improved Bioproduction in Corynebacterium glutamicum Irene Krahn1, Daniel Bonder2, Lucía Torregrosa-Barragán2, Dominik Stoppel1, 1 3 1 3,4 Edited by: Jens P. Krause , Natalie Rosenfeldt , Tobias M. Meiswinkel , Gerd M. Seibold , Pablo Ivan Nikel, Volker F. Wendisch1 and Steffen N. Lindner1,2* Novo Nordisk Foundation Center 1 2 for Biosustainability (DTU Biosustain), Chair of Genetics of Prokaryotes, Faculty of Biology and CeBiTec, Bielefeld University, Bielefeld, Germany, Systems 3 Denmark and Synthetic Metabolism, Max Planck Institute of Molecular Plant Physiology, Potsdam-Golm, Germany, Institute of Biochemistry, University of Cologne, Cologne, Germany, 4 Department of Biotechnology and Biomedicine, Technical Reviewed by: University of Denmark, Lyngby, Denmark Stephan Noack, Julich-Forschungszentrum, Helmholtz-Verband Deutscher Fructose utilization in Corynebacterium glutamicum starts with its uptake and Forschungszentren (HZ), Germany concomitant phosphorylation via the phosphotransferase system (PTS) to yield Fabien Létisse, UMR 5504 Laboratoire d’Ingénierie intracellular fructose 1-phosphate, which enters glycolysis upon ATP-dependent des Systèmes Biologiques et des phosphorylation to fructose 1,6-bisphosphate by 1-phosphofructokinase. This is known Procédés (LISBP), France to result in a significantly reduced oxidative pentose phosphate pathway (oxPPP) flux *Correspondence: ∼ ∼ Steffen N. Lindner on fructose ( 10%) compared to glucose ( 60%). Consequently, the biosynthesis of [email protected] NADPH demanding products, e.g., L-lysine, by C. glutamicum is largely decreased when fructose is the only carbon source. Previous works reported that fructose Specialty section: This article was submitted to is partially utilized via the glucose-specific PTS presumably generating fructose 6- Synthetic Biology, phosphate. -

Development and Control of Intestinal and Hepatic Fructokinase
INTESTINAL AND HEPATIC FRUCTOKINASE 765 expansion on intrarenal distribution of plasma flow in the dog. 42. The authors acknowledge the expert assistance of Mrs. Ann- Amer. J. Physiol., 223: 125 (1972). Christine Eklof and Mr. Gothe Carlebjork. 37. West, G. R., Smith, H. W., and Chasis, H.: Glomerular filtration 43. This research was supported by Grant no. B74-19X-3644-03Bfrom rate, effective renal blood flow and maximal tubular excretory the Swedish Medical Research Council and by a grant from the capacity in infancy. J. Pediat., 32: 10 (1948). Prenatal Research Funds of Expressen. 38. Laevosan Gesellschaft, Linz, Austria. 44. Requests for reprints should be addressed to: P. Herin, M.D., 39. 3M Company, St. Paul, Minn. Karolinski Institutet, Pediatriska Kliniken, St: Goran's Barn- 40. Sage Instruments, Inc., White Plains, N. Y. liniker, Box 12500, 112 81 Stockholm, Sweden. 41. Gilford Instrument Labs., Inc., Oberlin, Ohio. 45. Accepted for publication April 4, 1974. Copyright O 1974 International Pediatric Research Foundation, Inc. Printed in U.S.A. Pediat. Res. 8: 765-770 (1974) Developmental biochemistry intestine fetus liver fruct okinase Development and Control of Intestinal and Hepatic Fructokinase RICHARD J. GRAND,(^') MARIA I. SCHAY, AND STEPHANIE JAKSINA Department of Pediatrics, Harvard Medical School, and the Department of Medicine (Division of Clinical Nutrition), the Children's Hospital Medical Center, Boston, Massachusetts, USA Extract experimental animals (1, 2, 20, 26) and in man (I 5), although the mechanism of this regulation has not yet been elucidated. The patterns of development of intestinal and hepatic In the rat, adrenalectomy prevents the rise in the activity of fructokinase have been studied in the rat, in the rabbit, and in the hepatic enzyme expected after fructose feeding; hydro- man, and information regarding the mechanism of control of cortisone administered to the intact animal greatly enhances this enzyme in mature organs has been obtained. -

Cofermentation of Glucose, Xylose and Arabinose by Genomic DNA
GenomicCopyright © DNA–Integrated2002 by Humana Press Zymomonas Inc. mobilis 885 All rights of any nature whatsoever reserved. 0273-2289/02/98-100/0885/$13.50 Cofermentation of Glucose, Xylose, and Arabinose by Genomic DNA–Integrated Xylose/Arabinose Fermenting Strain of Zymomonas mobilis AX101 ALI MOHAGHEGHI,* KENT EVANS, YAT-CHEN CHOU, AND MIN ZHANG Biotechnology Division for Fuels and Chemicals, National Renewable Energy Laboratory, 1617 Cole Boulevard, Golden, CO 80401, E-mail: [email protected] Abstract Cofermentation of glucose, xylose, and arabinose is critical for complete bioconversion of lignocellulosic biomass, such as agricultural residues and herbaceous energy crops, to ethanol. We have previously developed a plas- mid-bearing strain of Zymomonas mobilis (206C[pZB301]) capable of cofer- menting glucose, xylose, and arabinose to ethanol. To enhance its genetic stability, several genomic DNA–integrated strains of Z. mobilis have been developed through the insertion of all seven genes necessay for xylose and arabinose fermentation into the Zymomonas genome. From all the integrants developed, four were selected for further evaluation. The integrants were tested for stability by repeated transfer in a nonselective medium (containing only glucose). Based on the stability test, one of the integrants (AX101) was selected for further evaluation. A series of batch and continuous fermenta- tions was designed to evaluate the cofermentation of glucose, xylose, and L-arabinose with the strain AX101. The pH range of study was 4.5, 5.0, and 5.5 at 30°C. The cofermentation process yield was about 84%, which is about the same as that of plasmid-bearing strain 206C(pZB301). Although cofer- mentation of all three sugars was achieved, there was a preferential order of sugar utilization: glucose first, then xylose, and arabinose last. -

Production of Alcohol for Fuel and Organic Solvents - M
BIOTECHNOLOGY – Vol. VII - Production of Alcohol for Fuel and Organic Solvents - M. Bekers and A. Vigants PRODUCTION OF ALCOHOL FOR FUEL AND ORGANIC SOLVENTS M. Bekers and A. Vigants Institute of Microbiology and Biotechnology, University of Latvia, Riga, Latvia Keywords: fuel ethanol, yeasts, organic solvents, biomass, liquefaction, saccharification, fermentation Contents 1. Production of Fuel Ethanol 1.1. Introduction 1.1.1. History and Actuality 1.1.2. Environmental Effects of Fuel Ethanol 1.1.3. Economic Aspects and Government Policies Affecting Fuel Ethanol Production and Marketing 1.2. Background 1.2.1. Producers and Regulation of Metabolism of Ethanol Synthesis 1.2.2. Raw Materials—Renewable Resources 1.3. Industrial Technologies 1.3.1. Medium Preparation 1.3.2. Fermentation 1.3.3. Distillation and Waste Utilization 1.4. Laboratory and Pilot Scale Technologies 1.4.1. Ethanol Production by the Bacterium Zymomonas mobilis 1.4.2. Immobilized, Flocculating and Recycling Systems 1.4.3. Vacuum and Extractive Fermentation 1.4.4. CASH Process and the Fermentation of Cellulosic Hydrolysates 2. Production of Organic Solvents 2.1. Introduction 2.1.1. History 2.1.2. Use and Properties of Solvents 2.2. Background 2.3. Technology 2.3.1. BatchUNESCO Process – EOLSS 2.3.2. Semicontinuous Process 2.3.3. Continuous Process 2.3.4. ImprovementsSAMPLE of Technology CHAPTERS 2.3.5. Utilization of Stillage 3. Conclusion Glossary Bibliography Biographical Sketches Summary The presented article describes the production of fuel ethanol and organic solvent ©Encyclopedia of Life Support Systems (EOLSS) BIOTECHNOLOGY – Vol. VII - Production of Alcohol for Fuel and Organic Solvents - M. -

Regulation of Fructose-6-Phosphate 2-Kinase By
Proc. Natt Acad. Sci. USA Vol. 79, pp. 325-329, January 1982 Biochemistry Regulation of fructose-6-phosphate 2-kinase by phosphorylation and dephosphorylation: Possible mechanism for coordinated control of glycolysis and glycogenolysis (phosphofructokinase) EISUKE FURUYA*, MOTOKO YOKOYAMA, AND KOSAKU UYEDAt Pre-Clinical Science Unit of the Veterans Administration Medical Center, 4500 South Lancaster Road, Dallas, Texas 75216; and Biochemistry Department of the University ofTexas Health Science Center, 5323 Harry Hines Boulevard, Dallas, Texas 75235 Communicated by Jesse C. Rabinowitz, September 28, 1981 ABSTRACT The kinetic properties and the control mecha- Fructose 6-phosphate + ATP nism of fructose-6-phosphate 2-kinase (ATP: D-fructose-6-phos- -3 Fructose + ADP. [1] phate 2-phosphotransferase) were investigated. The molecular 2,6-bisphosphate weight of the enzyme is -100,000 as determined by gel filtration. The plot of initial velocity versus ATP concentration is hyperbolic We have shown that the administration of extremely low con- with a K. of 1.2 mM. However, the plot of enzyme activity as a centrations of glucagon (0.1 fM) or high concentrations of epi- function of fructose 6-phosphate is sigmoidal. The apparent K0.5 nephrine (10 ,uM) to hepatocytes results in inactivation offruc- for fructose 6-phosphate is 20 ,IM. Fructose-6-phosphate 2-kinase tose-6-phosphate 2-kinase and concomitant decrease in the is inactivated by -the catalytic subunit of cyclic AMP-dependent fructose 2,6-bisphosphate level (12). These results, as well as protein kinase, and the inactivation is closely correlated with phos- more recent data using Ca2+ and the Ca2+ ionophore A23187 phorylation.