Trifluoromethanesulfonic Anhydride in Amide Activation:A Powerful Tool to Forge Heterocyclic Cores |
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -
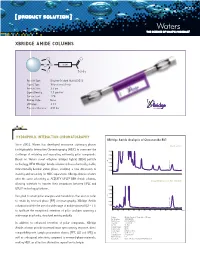
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

Detection of Atmospheric Gaseous Amines and Amides by a High-Resolution Time-Of-flight Chemical Ionization Mass Spectrometer with Protonated Ethanol Reagent Ions
Atmos. Chem. Phys., 16, 14527–14543, 2016 www.atmos-chem-phys.net/16/14527/2016/ doi:10.5194/acp-16-14527-2016 © Author(s) 2016. CC Attribution 3.0 License. Detection of atmospheric gaseous amines and amides by a high-resolution time-of-flight chemical ionization mass spectrometer with protonated ethanol reagent ions Lei Yao1, Ming-Yi Wang1,a, Xin-Ke Wang1, Yi-Jun Liu1,b, Hang-Fei Chen1, Jun Zheng2, Wei Nie3,4, Ai-Jun Ding3,4, Fu-Hai Geng5, Dong-Fang Wang6, Jian-Min Chen1, Douglas R. Worsnop7, and Lin Wang1,4 1Shanghai Key Laboratory of Atmospheric Particle Pollution and Prevention (LAP3), Department of Environmental Science & Engineering, Fudan University, Shanghai 200433, China 2Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing University of Information Science & Technology, Nanjing 210044, China 3Joint International Research Laboratory of Atmospheric and Earth System Sciences, School of Atmospheric Science, Nanjing University, Nanjing 210023, China 4Collaborative Innovation Center of Climate Change, Nanjing 210023, China 5Shanghai Meteorology Bureau, Shanghai 200135, China 6Shanghai Environmental Monitoring Center, Shanghai 200030, China 7Aerodyne Research, Billerica, MA 01821, USA anow at: Center for Atmospheric Particle Studies, Carnegie Mellon University, Pittsburgh, PA 15213, USA bnow at: Pratt School of Engineering, Duke University, Durham, NC 27705, USA Correspondence to: Lin Wang([email protected]) Received: 7 June 2016 – Published in Atmos. Chem. Phys. Discuss.: 22 June 2016 Revised: 15 October 2016 – Accepted: 3 November 2016 – Published: 23 November 2016 Abstract. Amines and amides are important atmospheric concentrations of amines ranged from a few parts per trillion organic-nitrogen compounds but high time resolution, highly by volume to hundreds of parts per trillion by volume, con- sensitive, and simultaneous ambient measurements of these centrations of amides varied from tens of parts per trillion by species are rather sparse. -

Downloaded 9/24/2021 4:45:01 AM
ORGANIC CHEMISTRY FRONTIERS View Article Online REVIEW View Journal | View Issue Recent developments in dehydration of primary amides to nitriles Cite this: Org. Chem. Front., 2020, 7, 3792 Muthupandian Ganesan *a and Paramathevar Nagaraaj *b Dehydration of amides is an efficient, clean and fundamental route for the syntheses of nitriles in organic chemistry. The two imperative functional groups viz., amide and nitrile groups have been extensively dis- cussed in the literature. However the recent development in the century-old dehydration method for the conversion of amides to nitriles has hardly been reported in one place, except a lone review article which dealt with only metal catalysed conversions. The present review provides broad and rapid information on the different methods available for the nitrile synthesis through dehydration of amides. The review article Received 15th July 2020, has major focus on (i) non-catalyzed dehydrations using chemical reagents, and (ii) catalyzed dehy- Accepted 19th September 2020 drations of amides using transition metal, non-transition metal, organo- and photo-catalysts to form the DOI: 10.1039/d0qo00843e corresponding nitriles. Also, catalyzed dehydrations in the presence of acetonitrile and silyl compounds as rsc.li/frontiers-organic dehydrating agents are highlighted. 1. Introduction polymers, materials, etc.1 Examples of pharmaceuticals contain- ing nitrile groups include vildagliptin, an anti-diabetic drug2 Nitriles are naturally found in various bacteria, fungi, plants and anastrazole, a drug -

First Principle Analysis on Pyridine Amide Derivatives' Adsorption Behavior on the Pt (111) Surface
crystals Article First Principle Analysis on Pyridine Amide Derivatives’ Adsorption Behavior on the Pt (111) Surface Guocai Tian *, Huanhuan Du and Hongmei Zhang Faculty of Metallurgical and Energy Engineering, State Key Laboratory of Complex Non-Ferrous Metal Resource Clean Utilization, Kunming University of Science and Technology, Kunming 650093, China; [email protected] (H.D.); [email protected] (H.Z.) * Correspondence: [email protected]; Tel.: +86-871-6519-8154 Abstract: The reactivity and adsorption behavior of three pyridine amide additives (Nicotinamide, Pyridine-2-formamide and Pyridine-4-formamide) on the Pt (111) surface was studied by First principle methods. The quantum chemical calculations of molecular reactivity show that the frontier orbitals of the three additives are distributed around the pyridine ring, oxygen atom of carbonyl and nitrogen atom of amino, and the nucleophilic and electrophilic active centers are located on the nitrogen atoms of pyridine ring, oxygen atom of carbonyl and nitrogen atom of amino. All three molecules were adsorbed with the chemical adsorption on the Pt (111) surface, and the order of adsorption was Nicotinamide > Pyridine-2-formamide > Pyridine-4-formamide. The C and N atoms of three derivatives forms C-Pt and N-Pt bonds with the Pt atoms of the Pt (111) surface, which makes derivatives stably adsorb on the Pt surface and form a protective film. The protective film inhibits the diffusion of atoms to the surface of the growth center, so as to inhibit the formation of dendrite and obtain a smooth aluminum deposition layer. Keywords: adsorption behavior; pyridine amide additives; first principle; molecular reactivity; aluminum deposition; chemical adsorption; Pt surface Citation: Tian, G.; Du, H.; Zhang, H. -

Organic Chemistry Test for Amide Group
Chemistry Organic Chemistry Test for Amide Group General Aim Method To test for amide-containing organic compounds Detection of the presence of amides using special through the chemical detection of amide groups. and specic reagents. Learning Objectives (ILOs) Dene and determine organic compounds containing amide groups theoretically through their chemical structure. Classify organic compounds containing amide groups into aliphatic and aromatic. Compare between amide groups and other functional groups in terms of chemical structures, properties and reactions. Identify amides experimentally. Select the appropriate reagents to dierentiate between amides and other organic compounds. Theoretical Background/Context - Amides are organic compounds that are considered as acid amides. Although the main dominant class of amides is the carboxyamide, phosphamides and sulphamides are also abundant. - Carboxyamindes possess at least one amide group (-CONH-).The functional group a carbonyl group (C=O) and amino group (-NH2) linked together. Amides (R – CON – R2’) are characterized by the possibility of having its R groups as a terminal H in their structure. In other words, the N can be linked with one or two H atoms instead of R groups. - The role that amides play in our real life is controlled by their properties. Amides are used in various applications in dierent elds such as food industry, pharmaceutics, etc. First: Preparation of Amides The simplest form of amide is prepared from ammonia, where a hydrogen atom is replaced by an acyl group. The product is described as a primary amide where its general formula is represented as R – CO – NH2. In the same way, secondary and tertiary amides can be prepared from primary and secondary amines, respectively. -
Chapter 6 Amines and Amides
Chapter 6 Amines and Amides Chapter 6 Amines and Amides Chapter Objectives: • Learn to recognize the amine and amide functional groups. • Learn the IUPAC system for naming amines and amides. • Learn the important physical properties of the amines and amides. • Learn the major chemical reactions of amines and amides, and learn how to predict the products of amide synthesis and hydrolysis reactions. • Learn some of the important properties of condensation polymers, especially the polyamides. Mr. Kevin A. Boudreaux Angelo State University CHEM 2353 Fundamentals of Organic Chemistry Organic and Biochemistry for Today (Seager & Slabaugh) www.angelo.edu/faculty/kboudrea Nitrogen-Containing Functional Groups • Nitrogen is in Group V of the periodic table, and in most of its compounds, it has three single bonds and one lone pair: N • In this chapter, we will take a look at two functional groups which contain nitrogen atoms connected to carbons: the amines and the amides. O RR''N RCN R' R' R" Amine Amide 2 Chapter 6 Amines and Amides Classification and Nomenclature of Amines 3 Amines • Amines and amides are abundant in nature. They are a major component of proteins and enzymes, nucleic acids, alkaloid drugs, etc. (Alkaloids are N- containing, weakly basic organic compounds; thousands of these substances are known.) • Amines are organic derivatives of ammonia, NH3, in which one or more of the three H’s is replaced by a carbon group. • Amines are classified as primary (1°), secondary (2°), or tertiary (3°), depending on how many carbon groups are connected to the nitrogen atom. HHN RHN RHN RR''N H H R' R' Ammonia 1° Amine 2° Amine 3° Amine 4 Chapter 6 Amines and Amides Examples: Classifying Amines • Classify the following amines as primary (1°), secondary (2°), or tertiary (3°). -

1 CHE 3332 – Organic Chemistry 2 – Spring 2021 Week 12 Resource
CHE 3332 – Organic Chemistry 2 – Spring 2021 Week 12 Resource Hello everyone! Virtual group tutoring for this class will be every Tuesday from 6-7 pm, and you can reserve your spot by making an appointment at www.Baylor.edu/Tutoring. This week, we’ll cover Chapter 21: Carboxylic Acid Derivatives. Keywords: carboxylic acid, acid halide, ester, amide, nitrile, anhydride Carboxylic Acids These are the reactions that make carboxylic acids (review from previous chapters): 1. Ozonolysis of alkyne 2. Oxidation of primary alcohol 3. Oxidation at benzylic position 4. Hydrolysis of nitrile 5. Carboxylation of Grignard Reactions that start with carboxylic acid: 1. Reduction with LAH or BH3. LAH is very powerful and reduces everything; BH3 is more selective and only reduces the carboxylic acid substituent. 2. Substitution – proton transfers can happen at any step 1 Acid Halides Reaction that makes acid halide with SOCl2 or COCl2 (chair lift reaction!): Reactions that start with acid halide: 1. Make a carboxylic acid 2. Make an ester – acyl substitution 3. Make an amide 4. Reduction to alcohol or aldehyde 5. Make an anhydride 6. React with Grignard or Gilman Acid Anhydrides Reactions that make acid anhydride: 1. From carboxylic acid 2. From acid halide as shown on page 2. 2 Esters Reactions that make esters: 1. From a carboxylic acid – SN2 2. Fischer Esterification 3. From acid halide as shown on page 2. Reactions that start with esters: 1. Transesterification – an ester turns into a new ester 2. Make a carboxylic acid with acid or base catalyzed reagents 3. Make an amide 4. -

Chapter 16 – Amines and Amides
Chapter 16 – Amines and Amides 16.1 Occurrence, Names, and Physical Properties of Amines An amine is basically an ammonia molecule with one or more aliphatic and/or aromatic organic groups attached. Thus they have the generic formulas NH2R, NHR2, and NR3. If a carbonyl group lies between the nitrogen and R the compound is called an amide. Amines are typically named by naming the organic group as a substituent and using the suffix “-amine.” This is the old common system and we will use it for the rest of the course. If an -NH2 group is named as a substituent, it goes by “amino-”. For example: O CH3NH2 (CH3)2NH (CH3)2N(C2H5) CH3CNH2 acetamide methylamine dimethylamine ethyldimethylamine ethanamide Aromatic amines are called anilines. When other groups are also attached to the nitrogen in aniline, they are named as for amines, except that a capital “N” is placed in front to indicate the substituent group is bound to the nitrogen, rather than the aromatic ring. A couple of examples include: NH2 N(CH3)2 aniline N,N-dimethylaniline Aromatic amides are analogous to their aliphatic counterparts. Two amines/amides that you have heard of are: H O O N HO N N n H O H Acetaminophen Nylon-66 N-aceto-4-aminophenol poly(hexamethylene adipamide) 2 Many natural products chemicals (these are species extracted from living systems) contain amino groups. The alkaloids are a class of these materials. If you see a drug end in “-ine” there is a good chance of it containing an amino group. Examples include: codeine, morphine, and quinine (all have pictures in your book on p. -

1 Heteroatom Substitution at Amide Nitrogen — Resonance Reduction and HERON 2 Reactions of Anomeric Amides 3 4 Stephen A
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 8 October 2018 doi:10.20944/preprints201810.0128.v1 Peer-reviewed version available at Molecules 2018, 23, 2834; doi:10.3390/molecules23112834 1 Heteroatom Substitution at Amide Nitrogen — Resonance Reduction and HERON 2 Reactions of Anomeric Amides 3 4 Stephen A. Glover and Adam A. Rosser 5 Department of Chemistry, 6 School of Science and Technology, 7 University of New England, Armidale, NSW 2351, Australia 8 Corresponding author, Email: [email protected] 9 10 Abstract: This review describes how resonance in amides is greatly affected upon substitution 11 at nitrogen by two electronegative atoms. Nitrogen becomes strongly pyramidal and resonance 12 stabilisation, evaluated computationally, can be reduced to as little as 50% that of N,N- 13 dimethylacetamide. However, this occurs without significant twisting about the amide bond, 14 which is borne out both experimentally and theoretically. In certain configurations, reduced 15 resonance and pronounced anomeric effects between heteroatom substituents are instrumental 16 in driving the HERON (Heteroatom Rearrangement On Nitrogen)† reaction, in which the more 17 electronegative atom migrates from nitrogen to the carbonyl carbon in concert with heterolysis 18 of the amide bond, to generate acyl derivatives and heteroatom-substituted nitrenes. In other 19 cases the anomeric effect facilitates SN1 and SN2 reactivity at the amide nitrogen. 20 21 1. Introduction 22 Amides are prevalent in a range of molecules such as peptides, proteins, lactams and many 23 synthetic polymers [1]. Generically, they are composed of both a carbonyl and an amino 24 functional group, joined by a single bond between the carbon and nitrogen. -
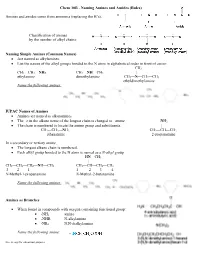
Naming Amines and Amides (Rules)
Chem 30B - Naming Amines and Amides (Rules) Amines and amides come from ammonia (replacing the H’s). Classification of amines by the number of alkyl chains: Naming Simple Amines (Common Names) • Are named as alkylamines. • List the names of the alkyl groups bonded to the N atom in alphabetical order in front of amine. CH3 CH3—CH2—NH2 CH3—NH—CH3 | ethylamine dimethylamine CH3—N—CH2—CH3 ethyldimethylamine Name the following amines: IUPAC Names of Amines • Amines are named as alkanamines. • The –e in the alkane name of the longest chain is changed to –amine. NH2 • The chain is numbered to locate the amine group and substituents. | CH3—CH2—NH2 CH3—CH—CH3 ethanamine 2-propanamine In a secondary or tertiary amine, • The longest alkane chain is numbered. • Each alkyl group bonded to the N atom is named as a N-alkyl group HN—CH3 | CH3—CH2—CH2—NH—CH3 CH3—CH—CH2—CH3 3 2 1 1 2 3 4 N-Methyl-1-propanamine N-Methyl-2-butanamine Name the following amines: Amines as Branches • When found in compounds with oxygen containing functional group: • -NH2 amino • -NHR N-alkylamino • -NR2 N,N-dialkylamino Name the following amine: Free to copy for educational purposes Ammonium Salts • Named by naming the positive ion first and then naming the negative ion. • Pharmaceutical companies often name amine salts by naming the parent amine followed by the name of the acid used to synthesize the salt: • HCl: amine hydrochlorides • H2SO4: amine hydrogen sulfates Amides Classification of amides by the number of alkyl chains: Amides are named as alkanamides.