1 Heteroatom Substitution at Amide Nitrogen — Resonance Reduction and HERON 2 Reactions of Anomeric Amides 3 4 Stephen A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organocatalytic Asymmetric N-Sulfonyl Amide C-N Bond Activation to Access Axially Chiral Biaryl Amino Acids
ARTICLE https://doi.org/10.1038/s41467-020-14799-8 OPEN Organocatalytic asymmetric N-sulfonyl amide C-N bond activation to access axially chiral biaryl amino acids Guanjie Wang1, Qianqian Shi2, Wanyao Hu1, Tao Chen1, Yingying Guo1, Zhouli Hu1, Minghua Gong1, ✉ ✉ ✉ Jingcheng Guo1, Donghui Wei 2 , Zhenqian Fu 1,3 & Wei Huang1,3 1234567890():,; Amides are among the most fundamental functional groups and essential structural units, widely used in chemistry, biochemistry and material science. Amide synthesis and trans- formations is a topic of continuous interest in organic chemistry. However, direct catalytic asymmetric activation of amide C-N bonds still remains a long-standing challenge due to high stability of amide linkages. Herein, we describe an organocatalytic asymmetric amide C-N bonds cleavage of N-sulfonyl biaryl lactams under mild conditions, developing a general and practical method for atroposelective construction of axially chiral biaryl amino acids. A structurally diverse set of axially chiral biaryl amino acids are obtained in high yields with excellent enantioselectivities. Moreover, a variety of axially chiral unsymmetrical biaryl organocatalysts are efficiently constructed from the resulting axially chiral biaryl amino acids by our present strategy, and show competitive outcomes in asymmetric reactions. 1 Key Laboratory of Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, China. 2 College -

779 Part 770—Interpretations
Pt. 770 15 CFR Ch. VII (1–1–21 Edition) in the item that qualitatively affect the per- roller bearings and parts). This applies formance of the U.S. and foreign items; to separate shipments of anti-friction (vi) Evidence of the interchangeability of bearings or bearing systems and anti- U.S. and foreign items; friction bearings or bearing systems (vii) Patent descriptions for the U.S. and foreign items; shipped with machinery or equipment (viii) Evidence that the U.S. and foreign for which they are intended to be used items meet a published industry, national, or as spares or replacement parts. international standard; (2) An anti-friction bearing or bear- (ix) A report or eyewitness account, by ing system physically incorporated in a deposition or otherwise, of the foreign item’s segment of a machine or in a complete operation; machine prior to shipment loses its (x) Evidence concerning the foreign manu- identity as a bearing. In this scenario, facturers’ corporate reputation; (xi) Comparison of the U.S. and foreign end the machine or segment of machinery item(s) made from a specific commodity, containing the bearing is the item sub- tool(s), device(s), or technical data; or ject to export control requirements. (xii) Evidence of the reputation of the for- (3) An anti-friction bearing or bear- eign item including, if possible, information ing system not incorporated in a seg- on maintenance, repair, performance, and ment of a machine prior to shipment, other pertinent factors. but shipped as a component of a com- plete unassembled (knocked-down) ma- SUPPLEMENT NO. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

Reliable Determination of Amidicity in Acyclic Amides and Lactams Stephen A
Article pubs.acs.org/joc Reliable Determination of Amidicity in Acyclic Amides and Lactams Stephen A. Glover* and Adam A. Rosser Department of Chemistry, School of Science and Technology, University of New England, Armidale, NSW 2351, Australia *S Supporting Information ABSTRACT: Two independent computational methods have been used for determination of amide resonance stabilization and amidicities relative to N,N-dimethylacetamide for a wide range of acyclic and cyclic amides. The first method utilizes carbonyl substitution nitrogen atom replacement (COSNAR). The second, new approach involves determination of the difference in amide resonance between N,N-dimethylacetamide and the target amide using an isodesmic trans-amidation process and is calibrated relative to 1-aza-2-adamantanone with zero amidicity and N,N-dimethylace- tamide with 100% amidicity. Results indicate excellent coherence between the methods, which must be regarded as more reliable than a recently reported approach to amidicities based upon enthalpies of hydrogenation. Data for acyclic planar and twisted amides are predictable on the basis of the degrees of pyramidalization at nitrogen and twisting about the C−N bonds. Monocyclic lactams are predicted to have amidicities at least as high as N,N-dimethylacetamide, and the β-lactam system is planar with greater amide resonance than that of N,N- dimethylacetamide. Bicyclic penam/em and cepham/em scaffolds lose some amidicity in line with the degree of strain-induced pyramidalization at the bridgehead nitrogen and twist about the amide bond, but the most puckered penem system still retains substantial amidicity equivalent to 73% that of N,N-dimethylacetamide. ■ INTRODUCTION completed by a third resonance form, III, with no C−N pi Amide linkages are ubiquitous in proteins, peptides, and natural character and on account of positive polarity at carbon and the or synthetic molecules and in most of these they possess, electronegativity of nitrogen, must be destabilizing. -

UNIT – I Nomenclature of Organic Compounds and Reaction Intermediates
UNIT – I Nomenclature of organic Compounds and Reaction Intermediates Two Marks 1. Give IUPAC name for the compounds. 2. Write the stability of Carbocations. 3. What are Carbenes? 4. How Carbanions reacts? Give examples 5. Write the reactions of free radicals 6. How singlet and triplet carbenes react? 7. How arynes are formed? 8. What are Nitrenium ion? 9. Give the structure of carbenes and Nitrenes 10. Write the structure of carbocations and carbanions. 11. Give the generation of carbenes and nitrenes. 12. What are non-classical carbocations? 13. How free radicals are formed and give its generation? 14. What is [1,2] shift? Five Marks 1. Explain the generation, Stability, Structure and reactivity of Nitrenes 2. Explain the generation, Stability, Structure and reactivity of Free Radicals 3. Write a short note on Non-Classical Carbocations. 4. Explain Fries rearrangement with mechanism. 5. Explain the reaction and mechanism of Sommelet-Hauser rearrangement. 6. Explain Favorskii rearrangement with mechanism. 7. Explain the mechanism of Hofmann rearrangement. Ten Marks 1. Explain brefly about generation, Stability, Structure and reactivity of Carbocations 2. Explain the generation, Stability, Structure and reactivity of Carbenes in detailed 3. Explain the generation, Stability, Structure and reactivity of Carboanions. 4. Explain brefly the mechanism and reaction of [1,2] shift. 5. Explain the mechanism of the following rearrangements i) Wolf rearrangement ii) Stevens rearrangement iii) Benzidine rearragement 6. Explain the reaction and mechanism of Dienone-Phenol reaarangement in detailed 7. Explain the reaction and mechanism of Baeyer-Villiger rearrangement in detailed Compounds classified as heterocyclic probably constitute the largest and most varied family of organic compounds. -

Transition Metal-Catalyzed Directed C(Sp3)–H Functionalization of Saturated Heterocycles
Synthesis Review / Short Review Transition Metal-Catalyzed Directed C(sp3)–H Functionalization of Saturated Heterocycles Daniele Antermitea James A. Bull*a a Department of Chemistry, Imperial College London, White City, Wood Lane, London, W12 0BZ, United Kingdom [email protected] Click here to insert a dedication. Received: biological interactions or selectivity profiles. The ready Accepted: Published online: availability of simple saturated heterocycle derivatives, DOI: including enantioenriched derivatives, makes them ideal Abstract Synthetic methods that can readily access saturated heterocycles starting points for further reactions. Therefore, approaches to with different substitution patterns and with control of stereo- and functionalize existing C–H bonds of these readily available regiochemistry are of huge potential value in the development of new medicinal compounds. Directed C–H functionalization of simple and building blocks appears to be of considerable potential. commercially available precursors offers the potential to prepare diverse Over the last 20 years, the concept of transition metal- collections of such valuable compounds that can probe the different available exit vectors from a ring system. Nonetheless, the presence of the catalyzed C–H functionalization has emerged with enormous Lewis basic heteroatoms makes this a significant challenge. This review potential to streamline the synthesis of complex molecules.5 covers recent advances in the catalytic C–H functionalization of saturated Specifically, transition metal catalysts can activate C–H bonds heterocycles, with a view to different heterocycles (N, O, S), substitution to form discrete C–M bonds, via different mechanistic patterns and transformations. 1. Introduction pathways.6 The resulting organometallic intermediate can then 2 a-C–H Functionalization with directing group on nitrogen form new C–C or C–heteroatom bonds with various coupling 3 C–H Functionalization at unactivated C(3), C(4) and C(5) positions partners. -

INFORMATION to USERS This Manuscript Has Been Reproduced
INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer primer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, prim bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g^ maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back of the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6” x 9” black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. A Bell & Howell Intormaiion Company 300 North Zeeb Road Ann Arbor Ml 46106*1346 USA 313/761-4700 800.521-0600 EXPLORATORY PHOTOCHEMISTRY OF POLYFLUORINATED ARYL AZIDES IN NEUTRAL AND ACIDIC MEDIA DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in the Graduate School of The Ohio State University By Hongbin Zhai ***** The Ohio State University 1995 Dissertation Committee: Approved By Matthew S. -

Electrochemistry and Photoredox Catalysis: a Comparative Evaluation in Organic Synthesis
molecules Review Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis Rik H. Verschueren and Wim M. De Borggraeve * Department of Chemistry, Molecular Design and Synthesis, KU Leuven, Celestijnenlaan 200F, box 2404, 3001 Leuven, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32-16-32-7693 Received: 30 March 2019; Accepted: 23 May 2019; Published: 5 June 2019 Abstract: This review provides an overview of synthetic transformations that have been performed by both electro- and photoredox catalysis. Both toolboxes are evaluated and compared in their ability to enable said transformations. Analogies and distinctions are formulated to obtain a better understanding in both research areas. This knowledge can be used to conceptualize new methodological strategies for either of both approaches starting from the other. It was attempted to extract key components that can be used as guidelines to refine, complement and innovate these two disciplines of organic synthesis. Keywords: electrosynthesis; electrocatalysis; photocatalysis; photochemistry; electron transfer; redox catalysis; radical chemistry; organic synthesis; green chemistry 1. Introduction Both electrochemistry as well as photoredox catalysis have gone through a recent renaissance, bringing forth a whole range of both improved and new transformations previously thought impossible. In their growth, inspiration was found in older established radical chemistry, as well as from cross-pollination between the two toolboxes. In scientific discussion, photoredox catalysis and electrochemistry are often mentioned alongside each other. Nonetheless, no review has attempted a comparative evaluation of both fields in organic synthesis. Both research areas use electrons as reagents to generate open-shell radical intermediates. Because of the similar modes of action, many transformations have been translated from electrochemical to photoredox methodology and vice versa. -

The Birth of Modern Chemistry
The Birth of Modern Chemistry 3rd semester project, Fall 2008 NIB Group nr.5, House 13.2 Thomas Allan Rayner & Aiga Mackevica Supervisor: Torben Brauner Abstract Alchemy was a science practiced for more than two millennia up till the end of 18th century when it was replaced by modern chemistry, which is practiced up till this very day. The purpose of this report is to look into this shift and investigate whether this shift can be classified as a paradigm shift according to the famous philosopher Thomas Kuhn, who came up with a theory on the structure of scientific revolutions. In order to come to draw any kind of conclusions, the report summarizes and defines the criteria for what constitutes a paradigm, crisis and paradigm shift, which are all important in order to investigate the manner in which a paradigm shift occurs. The criteria are applied to the historical development of modern chemistry and alchemy as well as the transition between the two sciences. As a result, alchemy and modern chemistry satisfy the requirements and can be viewed as two different paradigms in a Kuhnian sense. However, it is debatable whether the shift from alchemy to modern chemistry can be called a paradigm shift. 2 Acknowledgments We would like to thank Torben Brauner for his guidance throughout the project period as our supervisor. We would also like to thank our opposing group – Paula Melo Paulon Hansen, Stine Hesselholt Sloth and their supervisor Ole Andersen for their advice and critique for our project. 3 Table of Contents 1. Introduction ................................................................................................................................ 5 2. -
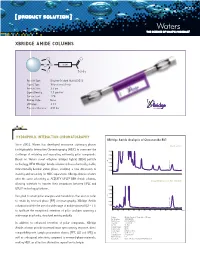
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj. -

General Chemistry Course Description
Chemistry 110: General Chemistry Note: this is a representative syllabus for Chemistry 110. It is here for informational purposes. It is not intended to substitute for or replace the syllabus your instructor provides. Course Description: Introduction to the general principles of chemistry for students planning a professional career in chemistry, a related science, the health professions, or engineering. Stoichiometry, atomic structure, chemical bonding and geometry, thermochemistry, gases, types of chemical reactions, statistics. Weekly laboratory exercises emphasize quantitative techniques and complement the lecture material. Weekly discussion sessions focus on homework assignments and lecture material. Prerequisite: Mathematics 055 or equivalent. Previous high school chemistry not required. Taught: Annually, 5 semester credits. Course Components: Lecture, Recitation section, Laboratory Textbook: Steven S. Zumdahl and Susan A. Zumdahl, Chemistry, 5th Ed., Houghton Mifflin Company (New York, 2000). The text is available in the college bookstore. Lab Manual: Chemistry Department Faculty, Chemistry 110 Lab Manual, Fall, 2001 Edition, Kinkos. These may be purchased from Linda Noble, the Chemistry Secretary. Safety Goggles: These must be worn at all times in the laboratory. They can be purchased in the college bookstore. Calculator: Must have logs and exponential capability. Lab Notebook: This is a bound duplicate notebook with numbered pages. It can be purchased in the college bookstore Lecture Schedule 1.1-1.5 introduction, laboratory precision -

Course Syllabus
Course Syllabus Department: Science & Technology Date: January 2015 I. Course Prefix and Number: CHM 205 Course Name: Organic Chemistry I – Lecture Only Credit Hours and Contact Hours: 4 credit hours and 4 (3:0:1) contact hours Catalog Description including pre- and co-requisites: A systematic study of the chemistry of carbon compounds emphasizing reactions, mechanisms, and synthesis with a focus on functional groups, addition reactions to alkenes and alkynes, alcohols and ethers, stereochemistry, nomenclature, acid-base chemistry, reaction kinetics and thermodynamics. Completion of General Chemistry II or equivalent with a grade of C or better is prerequisite. II. Course Outcomes and Objectives Student Learning Outcomes: Upon completion of this course, the student will be able to: Demonstrate an understanding of basic principles of organic chemistry and how they relate to everyday experiences. Demonstrate problem solving and critical thinking skills Apply methods of scientific inquiry. Apply problem solving techniques to real-world problems. Demonstrate an understanding of the chemical environment and the role that organic molecules play in the natural and the synthetic world. Relationship to Academic Programs and Curriculum: This course is required for majors in chemistry, chemical engineering, biology, biotechnology, pharmacology, and other pre-professional programs. College Learning Outcomes Addressed by the Course: writing computer literacy oral communications ethics/values X reading citizenship mathematics global concerns X critical thinking information resources 1 III. Instructional Materials and Methods Types of Course Materials: A standard two-semester 200 level organic textbook and workbook. Methods of Instruction (e.g. Lecture and Seminar …): Three hours of lecture, with a one hour recitation period for individual as well as group learning activities such as case studies and guided learning activities.