Reliable Determination of Amidicity in Acyclic Amides and Lactams Stephen A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organocatalytic Asymmetric N-Sulfonyl Amide C-N Bond Activation to Access Axially Chiral Biaryl Amino Acids
ARTICLE https://doi.org/10.1038/s41467-020-14799-8 OPEN Organocatalytic asymmetric N-sulfonyl amide C-N bond activation to access axially chiral biaryl amino acids Guanjie Wang1, Qianqian Shi2, Wanyao Hu1, Tao Chen1, Yingying Guo1, Zhouli Hu1, Minghua Gong1, ✉ ✉ ✉ Jingcheng Guo1, Donghui Wei 2 , Zhenqian Fu 1,3 & Wei Huang1,3 1234567890():,; Amides are among the most fundamental functional groups and essential structural units, widely used in chemistry, biochemistry and material science. Amide synthesis and trans- formations is a topic of continuous interest in organic chemistry. However, direct catalytic asymmetric activation of amide C-N bonds still remains a long-standing challenge due to high stability of amide linkages. Herein, we describe an organocatalytic asymmetric amide C-N bonds cleavage of N-sulfonyl biaryl lactams under mild conditions, developing a general and practical method for atroposelective construction of axially chiral biaryl amino acids. A structurally diverse set of axially chiral biaryl amino acids are obtained in high yields with excellent enantioselectivities. Moreover, a variety of axially chiral unsymmetrical biaryl organocatalysts are efficiently constructed from the resulting axially chiral biaryl amino acids by our present strategy, and show competitive outcomes in asymmetric reactions. 1 Key Laboratory of Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, China. 2 College -

The Birth of Modern Chemistry
The Birth of Modern Chemistry 3rd semester project, Fall 2008 NIB Group nr.5, House 13.2 Thomas Allan Rayner & Aiga Mackevica Supervisor: Torben Brauner Abstract Alchemy was a science practiced for more than two millennia up till the end of 18th century when it was replaced by modern chemistry, which is practiced up till this very day. The purpose of this report is to look into this shift and investigate whether this shift can be classified as a paradigm shift according to the famous philosopher Thomas Kuhn, who came up with a theory on the structure of scientific revolutions. In order to come to draw any kind of conclusions, the report summarizes and defines the criteria for what constitutes a paradigm, crisis and paradigm shift, which are all important in order to investigate the manner in which a paradigm shift occurs. The criteria are applied to the historical development of modern chemistry and alchemy as well as the transition between the two sciences. As a result, alchemy and modern chemistry satisfy the requirements and can be viewed as two different paradigms in a Kuhnian sense. However, it is debatable whether the shift from alchemy to modern chemistry can be called a paradigm shift. 2 Acknowledgments We would like to thank Torben Brauner for his guidance throughout the project period as our supervisor. We would also like to thank our opposing group – Paula Melo Paulon Hansen, Stine Hesselholt Sloth and their supervisor Ole Andersen for their advice and critique for our project. 3 Table of Contents 1. Introduction ................................................................................................................................ 5 2. -

General Chemistry Course Description
Chemistry 110: General Chemistry Note: this is a representative syllabus for Chemistry 110. It is here for informational purposes. It is not intended to substitute for or replace the syllabus your instructor provides. Course Description: Introduction to the general principles of chemistry for students planning a professional career in chemistry, a related science, the health professions, or engineering. Stoichiometry, atomic structure, chemical bonding and geometry, thermochemistry, gases, types of chemical reactions, statistics. Weekly laboratory exercises emphasize quantitative techniques and complement the lecture material. Weekly discussion sessions focus on homework assignments and lecture material. Prerequisite: Mathematics 055 or equivalent. Previous high school chemistry not required. Taught: Annually, 5 semester credits. Course Components: Lecture, Recitation section, Laboratory Textbook: Steven S. Zumdahl and Susan A. Zumdahl, Chemistry, 5th Ed., Houghton Mifflin Company (New York, 2000). The text is available in the college bookstore. Lab Manual: Chemistry Department Faculty, Chemistry 110 Lab Manual, Fall, 2001 Edition, Kinkos. These may be purchased from Linda Noble, the Chemistry Secretary. Safety Goggles: These must be worn at all times in the laboratory. They can be purchased in the college bookstore. Calculator: Must have logs and exponential capability. Lab Notebook: This is a bound duplicate notebook with numbered pages. It can be purchased in the college bookstore Lecture Schedule 1.1-1.5 introduction, laboratory precision -

Course Syllabus
Course Syllabus Department: Science & Technology Date: January 2015 I. Course Prefix and Number: CHM 205 Course Name: Organic Chemistry I – Lecture Only Credit Hours and Contact Hours: 4 credit hours and 4 (3:0:1) contact hours Catalog Description including pre- and co-requisites: A systematic study of the chemistry of carbon compounds emphasizing reactions, mechanisms, and synthesis with a focus on functional groups, addition reactions to alkenes and alkynes, alcohols and ethers, stereochemistry, nomenclature, acid-base chemistry, reaction kinetics and thermodynamics. Completion of General Chemistry II or equivalent with a grade of C or better is prerequisite. II. Course Outcomes and Objectives Student Learning Outcomes: Upon completion of this course, the student will be able to: Demonstrate an understanding of basic principles of organic chemistry and how they relate to everyday experiences. Demonstrate problem solving and critical thinking skills Apply methods of scientific inquiry. Apply problem solving techniques to real-world problems. Demonstrate an understanding of the chemical environment and the role that organic molecules play in the natural and the synthetic world. Relationship to Academic Programs and Curriculum: This course is required for majors in chemistry, chemical engineering, biology, biotechnology, pharmacology, and other pre-professional programs. College Learning Outcomes Addressed by the Course: writing computer literacy oral communications ethics/values X reading citizenship mathematics global concerns X critical thinking information resources 1 III. Instructional Materials and Methods Types of Course Materials: A standard two-semester 200 level organic textbook and workbook. Methods of Instruction (e.g. Lecture and Seminar …): Three hours of lecture, with a one hour recitation period for individual as well as group learning activities such as case studies and guided learning activities. -
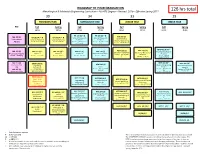
BS Metallurgical Engineering Curriculum Flowchart
ROADMAP TO YOUR GRADUATION Metallurgical & Materials Engineering Curriculum – BS MTE Degree – Revised 2016-- Effective Spring 2017 126 hrs total 30 34 33 29 FRESHMAN YEAR SOPHOMORE YEAR JUNIOR YEAR SENIOR YEAR Pre Fall SprIng Fall SprIng Fall SprIng Fall SprIng 16 hrs 14 hrs 17 hrs 17 hrs 16 hrs 17 hrs 14 hrs 15 hrs PH 105 (4) * N PH 106 (4) * N ECE 320 (3) MA 100 (3) CH 101 (4) * N CH 102 (4) * N General Physics with General Physics with Fundamentals of Intermediate General Chemistry 1 General Chemistry 2 Calculus 1 Calculus 2 Electrical Engineering Pre-reqs. = See catalog Pre-reqs. = CH 101 Pre-reqs. = MA 113 or 115 Pre-reqs. = PH 101 or 105 or Algebra Pre-reqs. = PH 106, MA 238 or 125 or 145 125 MTE 481 (4) W * AEM 250 (3) MTE 455 (4) MA 112 (3) MA 125 (4) * MA 126 (4) * MA 227 (4) MA 238 (3) Analytical Methods for Mechanics of Materials Mechanical Behavior of Precalculus Algebra Calculus 1 Calculus 2 Calculus 3 Differential Equations 1 Materials Pre-reqs. = MA 126, AEM Materials Pre-reqs. = See catalog Pre-reqs. = See catalog Pre-reqs. = MA 125 Pre-reqs. = MA 126 Pre-reqs. = MA 126 Pre-req. = MA 238 201 Pre-reqs. = AEM 250 Co-req. = MTE 441 # MA 113 (3) ENGR 103 (3) MTE 443 (3) MTE 445 (3) # AEM 201 (3) Materials Engineering Precalculus Trig Engineering Materials Engineering Statics Design 1 OR Foundations Design 2 Pre-reqs. = MA 125, PH 105, Pre-reqs. = MTE 362, 373, Pre-reqs. = MATH 125 or Pre-reqs. -

Professor of Chemistry
Bruce C. Gibb FRSC Professor of Chemistry Department of Chemistry Tulane University New Orleans, LA 70118, USA Tel: (504) 862 8136 E-mail: [email protected] Website: http://www.gibbgroup.org Twitter: @brucecgibb Research Interests: Aqueous supramolecular chemistry: understanding how molecules interact in water: from specific ion- pairing and the hydrophobic effect, to protein aggregation pertinent to neurodegenerative disorders. Our research has primarily focused on: 1) novel hosts designed to probe the hydrophobic, Hofmeister, and Reverse Hofmeister effects, and; 2) designing supramolecular capsules as yocto-liter reaction vessels and separators. Current efforts to probe the hydrophobic and Hofmeister effects include studies of the supramolecular properties of proteins. Professional Positions: Visiting Professor, Wuhan University of Science and Technology as a Chair Professor of Chutian Scholars Program (2015-2018) Professor of Chemistry, Tulane University, New Orleans, USA (2012-present). University Research Professor, University of New Orleans, USA (2007-2011). Professor of Chemistry, University of New Orleans, USA (2005-2007). Associate Professor of Chemistry, University of New Orleans, USA (2002-2005). Assistant Professor of Chemistry, University of New Orleans, USA, (1996-2002). Education: Postdoctoral Work Department of Chemistry, New York University. Synthesis of Carbonic Anhydrase (CA) mimics with Advisor: Prof. J. W. Canary, (1994-1996). Department of Chemistry, University of British Columbia, Canada De Novo Protein development. Advisor: Prof. J. C. Sherman (1993-1994). Ph.D. Robert Gordon’s University, Aberdeen, UK. Synthesis and Structural Examination of 3a,5-cyclo-5a- Androstane Steroids. Advisors: Dr. Philip J. Cox and Dr. Steven MacManus (1987-92) . B.Sc. with Honors in Physical Sciences Robert Gordon’s University, Aberdeen, UK. -

Chemistry (CH) 1
Chemistry (CH) 1 Chemistry (CH) CH 101 - INTRO TO CHEMISTRY Semester Hours: 3 Properties of solids, liquids, gases, and solutions, atomic theory and bonding, concentration concepts, and physical and chemical properties of the more common elements and their compounds. No placement examination is required. Prerequisite: MA 110 or prerequisites with concurrency MA 112 or higher and CH 105. CH 101R - RECITATION Semester Hours: 0 CH 105 - INTRO CHEMISTRY LAB Semester Hour: 1 Complements the lecture material for CH 101. Laboratory fundamentals and basic chemical principles. Prerequisite with concurrency: CH 101. CH 121 - GENERAL CHEMISTRY I Semester Hours: 3 For science and engineering majors. Chemical properties of elements, their periodic groups, and their compounds. Reactions and stoichiometry. Nature of the chemical bond, molecular structure, thermochemistry. Properties of gases, liquids, and solids. Prerequisite: CH 101 or placement test. Prerequisites with concurrency: MA 113 or higher, and CH 125. CH 121M - GENERAL CHEMISTRY FOR CHEMISTS Semester Hours: 3 For Chemistry Majors and Minors. Chemical properties of elements and the Periodic Table. Reactions and stoichiometry. Nature of chemical bonds, molecular structure, thermochemistry. Properties of gases, liquids, and solids. Intro to your exciting Department and its programs. Engagement activities. Prerequisites: CH 101 or Placement. Prerequisites with concurrency: MA 113 or higher, CH 125. CH 121R - RECITATION Semester Hours: 0 CH 122 - GENERAL CHEMISTRY ENGINEERS Semester Hours: 3 This course is designed as a one semester presentation of key aspects in general chemistry and is recommended for all engineering majors except chemical engineers. Covers topic on atoms and molecules: reactions and stoichiometry; gases; the periodic table; atomic structure, chemical bonding and molecular structure; materials; energy, entropy, and free energy; kinetics and equilibrium; and electrochemistry. -

Stoichiometry of Chemical Reactions 175
Chapter 4 Stoichiometry of Chemical Reactions 175 Chapter 4 Stoichiometry of Chemical Reactions Figure 4.1 Many modern rocket fuels are solid mixtures of substances combined in carefully measured amounts and ignited to yield a thrust-generating chemical reaction. (credit: modification of work by NASA) Chapter Outline 4.1 Writing and Balancing Chemical Equations 4.2 Classifying Chemical Reactions 4.3 Reaction Stoichiometry 4.4 Reaction Yields 4.5 Quantitative Chemical Analysis Introduction Solid-fuel rockets are a central feature in the world’s space exploration programs, including the new Space Launch System being developed by the National Aeronautics and Space Administration (NASA) to replace the retired Space Shuttle fleet (Figure 4.1). The engines of these rockets rely on carefully prepared solid mixtures of chemicals combined in precisely measured amounts. Igniting the mixture initiates a vigorous chemical reaction that rapidly generates large amounts of gaseous products. These gases are ejected from the rocket engine through its nozzle, providing the thrust needed to propel heavy payloads into space. Both the nature of this chemical reaction and the relationships between the amounts of the substances being consumed and produced by the reaction are critically important considerations that determine the success of the technology. This chapter will describe how to symbolize chemical reactions using chemical equations, how to classify some common chemical reactions by identifying patterns of reactivity, and how to determine the quantitative relations between the amounts of substances involved in chemical reactions—that is, the reaction stoichiometry. 176 Chapter 4 Stoichiometry of Chemical Reactions 4.1 Writing and Balancing Chemical Equations By the end of this section, you will be able to: • Derive chemical equations from narrative descriptions of chemical reactions. -

Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides
molecules Review Heteroatom Substitution at Amide Nitrogen—Resonance Reduction and HERON Reactions of Anomeric Amides Stephen A. Glover * and Adam A. Rosser Department of Chemistry, School of Science and Technology, University of New England, Armidale, NSW 2351, Australia; [email protected] * Correspondence: [email protected] Received: 6 October 2018; Accepted: 24 October 2018; Published: 31 October 2018 Abstract: This review describes how resonance in amides is greatly affected upon substitution at nitrogen by two electronegative atoms. Nitrogen becomes strongly pyramidal and resonance stabilisation, evaluated computationally, can be reduced to as little as 50% that of N,N-dimethylacetamide. However, this occurs without significant twisting about the amide bond, which is borne out both experimentally and theoretically. In certain configurations, reduced resonance and pronounced anomeric effects between heteroatom substituents are instrumental in driving the HERON (Heteroatom Rearrangement On Nitrogen) reaction, in which the more electronegative atom migrates from nitrogen to the carbonyl carbon in concert with heterolysis of the amide bond, to generate acyl derivatives and heteroatom-substituted nitrenes. In other cases the anomeric effect facilitates SN1 and SN2 reactivity at the amide nitrogen. Keywords: amide resonance; anomeric effect; HERON reaction; pyramidal amides; physical organic chemistry; reaction mechanism 1. Introduction Amides are prevalent in a range of molecules such as peptides, proteins, lactams, and many synthetic polymers [1]. Generically, they are composed of both a carbonyl and an amino functional group, joined by a single bond between the carbon and nitrogen. The contemporary understanding of the resonance interaction between the nitrogen and the carbonyl in amides is that of an interaction between the lowest unoccupied molecular orbital (LUMO) of the carbonyl, π*C=O, and the highest occupied molecular orbital (HOMO) of the amide nitrogen (N2pz) (Figure1). -

Contents Notes on General Chemistry, 2E
Contents Notes On General Chemistry, 2e Chapter 1, The stuff of chemistry .......................................................................... 1 Avogadro's hypothesis ................................................................................... 2 Incomplete combustion of acetylene .............................................................. 3 Building blocks of atoms ................................................................................ 3 Charge-to-mass ratio of the electron ........................................................... 3 Charge, and so mass of the electron ........................................................... 4 The nucleus ............................................................................................. 4 Protons, neutrons, and isotopes .................................................................. 6 Relative atomic mass ...................................................................................... 6 Absolute atomic mass ..................................................................................... 7 Calculating chemical amounts ........................................................................ 8 Given mass of an element, how many atoms do we have .............................. 8 Given mass of a compound, how many molecules do we have? .................... 9 Given mass of a compound, how many atoms do we have? .......................... 9 How much mass is needed to have a given number of molecules? ................. 10 How much mass is needed to have a given -

Chemistry (CHEM) 1
Chemistry (CHEM) 1 Chemistry (CHEM) CHEM 117. Chemical Concepts and Applications. 3 Credits. Introduction to general and organic chemistry, with applications drawn from the health, environmental, and materials sciences. Prereq or Coreq: MATH 103, MATH 104 or MATH 107 or Math placement. CHEM 117L. Chem Concepts and Applications Lab. 1 Credit. Introduction to general and organic chemistry, with applications drawn from the health, environmental, and materials sciences. Prereq or Coreq: MATH 103, MATH 104, MATH 107 or Math placement. CHEM 121L. General Chemistry I Laboratory. 1 Credit. Matter, measurement, atoms, ions, molecules, reactions, chemical calculations, thermochemistry, bonding, molecular geometry, periodicity, and gases. Prereq or Coreq: MATH 103 or MATH 107 or Math placement. CHEM 121. General Chemistry I. 3 Credits. Matter, measurement, atoms, ions, molecules, reactions, chemical calculations, thermochemistry, bonding, molecular geometry, periodicity, and gases. Prereq or Coreq: MATH 103 or MATH 107 or Math placement. CHEM 122L. General Chemistry II Laboratory. 1 Credit. Intermolecular forces, liquids, solids, kinetics, equilibria, acids and bases, solution chemistry, precipitation, thermodynamics, and electrochemistry. Prereq: CHEM 121L. CHEM 122. General Chemistry II. 3 Credits. Intermolecular forces, liquids, solids, kinetics, equilibria, acids and bases, solution chemistry, precipitation, thermodynamics, and electrochemistry. Prereq: CHEM 121. CHEM 140. Organic Chemical Concepts and Applications. 1 Credit. Introduction to organic chemistry for pre-nursing and other students who need to meet the prerequisite for CHEM 260. CHEM 150. Principles of Chemistry I. 3 Credits. Chemistry for students with good high school preparation in mathematics and science. Electronic structure, stoichiometry, molecular geometry, ionic and covalent bonding, energetics of chemical reactions, gases, transition metal chemistry. -

Introduction to General Chemistry Calculations and Concepts
Preparatory Chemistry: Introduction to General Chemistry Calculations and Concepts ABOZENADAH BISHOP BITTNER FLATT LOPEZ WILEY Published by Western Oregon University under Creative Commons Licensing as Attribution-NonCommercial-ShareAlike CC BY-NC-SA 3.0 on Dec 15th, 2017 To Share or Adapt this Content please Reference: Abozenadah, H., Bishop, A., Bittner, S.. Flatt, P.M., Lopez, O., and Wiley, C. (2017) Con- sumer Chemistry: How Organic Chemistry Impacts Our Lives. CC BY-NC-SA. Available at: Cover photo adapted from: https://upload.wikimedia.org/wikipedia/commons/thumb/e/e1/Chemicals_in_flasks. jpg/675px-Chemicals_in_flasks.jpg Chapter 1 materials have been adapted from the following creative commons resources unless otherwise noted: 1. Anonymous. (2012) Introduction to Chemistry: General, Organic, and Biological (V1.0). Published under Creative Commons by-nc-sa 3.0. Available at: http://2012books.lard- bucket.org/books/introduction-to-chemistry-general-organic-and-biological/index.html 2. Poulsen, T. (2010) Introduction to Chemistry. Published under Creative Commons by- nc-sa 3.0. Available at: http://openedgroup.org/books/Chemistry.pdf 3. OpenStax (2015) Atoms, Isotopes, Ions, and Molecules: The Building Blocks. OpenStax CNX.Available at: http://cnx.org/contents/be8818d0-2dba-4bf3-859a-737c25fb2c99@12. Table of Contents Section 1: Chemistry and Matter 4 What is Chemistry? 4 Physical vs. Chemical Properties 4 Elements and Compounds 5 Mixtures 7 States of Matter 8 Section 2: How Scientists Study Chemistry 10 The Scientific Method 10 Section 3: Scientific Notation 11 Section 4: Units of Measurement 14 International System of Units and the Metric System 14 Mass 16 Length 16 Temperature 17 Time 18 Amount 18 Derived SI Units 19 Volume 19 Energy 20 Density 20 Section 5: Making Measurements in the Lab 21 Precision vs.