Protein Cysteine Oxidation in Redox Signaling: Caveats on Sulfenic Acid Detection and Quantification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Norbornene Probes for the Detection of Cysteine Sulfenic Acid in Cells Lisa J
Letters Cite This: ACS Chem. Biol. 2019, 14, 594−598 pubs.acs.org/acschemicalbiology Norbornene Probes for the Detection of Cysteine Sulfenic Acid in Cells Lisa J. Alcock,†,‡ Bruno L. Oliveira,§ Michael J. Deery,∥ Tara L. Pukala,⊥ Michael V. Perkins,† Goncalo̧ J. L. Bernardes,*,§,# and Justin M. Chalker*,†,‡ † Flinders University, College of Science and Engineering, Sturt Road, Bedford Park, South Australia 5042, Australia ‡ Flinders University, Institute for NanoScale Science and Technology, Sturt Road, Bedford Park, South Australia 5042, Australia § Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, United Kingdom ∥ Cambridge Centre for Proteomics, Cambridge Systems Biology Centre, Department of Biochemistry, University of Cambridge, Tennis Court Road, Cambridge CB2 1QW, United Kingdom ⊥ The University of Adelaide, School of Physical Sciences, Adelaide, South Australia 5005, Australia # Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Avenida Professor Egas Moniz, 1649-028, Lisboa, Portugal *S Supporting Information ABSTRACT: Norbornene derivatives were validated as probes for cysteine sulfenic acid on proteins and in live cells. Trapping sulfenic acids with norbornene probes is highly selective and revealed a different reactivity profile than the traditional dimedone reagent. The norbornene probe also revealed a superior chemoselectivity when compared to a commonly used dimedone probe. Together, these results advance the study of cysteine oxidation in biological systems. -

Sodium Borohydride (Nabh4) Itself Is a Relatively Mild Reducing Agent
1770 Vol. 35 (1987) Chem. Pharm. Bull. _35( 5 )1770-1776(1987). Reactions of Sodium Borohydride. IV.1) Reduction of Aromatic Sulfonyl Chlorides with Sodium Borohydride ATSUKO NOSE and TADAHIRO KUDO* Daiichi College of Pharmaceutical Sciences, 22-1 Tamagawa-cho, Minami-ku, Fukuoka 815, Japan (Received September 12, 1986) Aromatic sulfonyl chlorides were reduced with sodium borohydride in tetrahydrofuran at 0•Ž to the corresponding sulfinic acids in good yields. Further reduction proceeded when the reaction was carried out under reflux in tetrahydrofuran to give disulfide and thiophenol derivatives via sulfinic acid. Furthermore, sulfonamides were reduced with sodium borohydride by heating directly to give sulfide, disulfide and thiophenol derivatives, and diphenyl sulfone was reduced under similar conditions to give thiophenol and biphenyl. Keywords reduction; sodium borohydride; aromatic sulfonyl chloride; sulfonamide; sulfone; aromatic sulfinic acid; disulfide; sulfide; thiophenol Sodium borohydride (NaBH4) itself is a relatively mild reducing agent which is extensively used for the selective reduction of the carbonyl group of ketone, aldehyde and (carboxylic) acid halide derivatives. In the previous papers, we reported that NaBI-14 can reduce some functional groups, such as carboxylic anhydrides,2) carboxylic acids3) and nitro compounds.1) As a continuation of these studies, in the present paper, we wish to report the reduction of aromatic sulfonyl chlorides, sulfinic acids and sulfonamides with NaBH4. Many methods have been reported for the reduction of sulfonyl chlorides to sulfinic acids, such as the use of sodium sulfite,4a-d) zinc,5) electrolytic reduction,6) catalytic reduction,7) magnesium,8) sodium amalgam,9) sodium hydrogen sulfite,10) stannous chlo- TABLE I. -

Amide Activation: an Emerging Tool for Chemoselective Synthesis
Featuring work from the research group of Professor As featured in: Nuno Maulide, University of Vienna, Vienna, Austria Amide activation: an emerging tool for chemoselective synthesis Let them stand out of the crowd – Amide activation enables the chemoselective modification of a large variety of molecules while leaving many other functional groups untouched, making it attractive for the synthesis of sophisticated targets. This issue features a review on this emerging field and its application in total synthesis. See Nuno Maulide et al., Chem. Soc. Rev., 2018, 47, 7899. rsc.li/chem-soc-rev Registered charity number: 207890 Chem Soc Rev View Article Online REVIEW ARTICLE View Journal | View Issue Amide activation: an emerging tool for chemoselective synthesis Cite this: Chem. Soc. Rev., 2018, 47,7899 Daniel Kaiser, Adriano Bauer, Miran Lemmerer and Nuno Maulide * It is textbook knowledge that carboxamides benefit from increased stabilisation of the electrophilic carbonyl carbon when compared to other carbonyl and carboxyl derivatives. This results in a considerably reduced reactivity towards nucleophiles. Accordingly, a perception has been developed of amides as significantly less useful functional handles than their ester and acid chloride counterparts. Received 27th April 2018 However, a significant body of research on the selective activation of amides to achieve powerful DOI: 10.1039/c8cs00335a transformations under mild conditions has emerged over the past decades. This review article aims at placing electrophilic amide activation in both a historical context and in that of natural product rsc.li/chem-soc-rev synthesis, highlighting the synthetic applications and the potential of this approach. Creative Commons Attribution 3.0 Unported Licence. -

8.6 Acidity of Alcohols and Thiols 355
08_BRCLoudon_pgs5-1.qxd 12/8/08 11:05 AM Page 355 8.6 ACIDITY OF ALCOHOLS AND THIOLS 355 ural barrier to the passage of ions. However, the hydrocarbon surface of nonactin allows it to enter readily into, and pass through, membranes. Because nonactin binds and thus transports ions, the ion balance crucial to proper cell function is upset, and the cell dies. Ion Channels Ion channels, or “ion gates,” provide passageways for ions into and out of cells. (Recall that ions are not soluble in membrane phospholipids.) The flow of ions is essen- tial for the transmission of nerve impulses and for other biological processes. A typical chan- nel is a large protein molecule imbedded in a cell membrane. Through various mechanisms, ion channels can be opened or closed to regulate the concentration of ions in the interior of the cell. Ions do not diffuse passively through an open channel; rather, an open channel contains regions that bind a specific ion. Such an ion is bound specifically within the channel at one side of the membrane and is somehow expelled from the channel on the other side. Remark- ably, the structures of the ion-binding regions of these channels have much in common with the structures of ionophores such as nonactin. The first X-ray crystal structure of a potassium- ion channel was determined in 1998 by a team of scientists at Rockefeller University led by Prof. Roderick MacKinnon (b. 1956), who shared the 2003 Nobel Prize in Chemistry for this work. The interior of the channel contains binding sites for two potassium ions; these sites are oxygen-rich, much like the interior of nonactin. -

N* Interactions Modulate the Properties of Cysteine Residues
n →π* Interactions Modulate the Properties of Cysteine Residues and Disulfide Bonds in Proteins The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Kilgore, Henry R. and Ronald T. Raines. “n →π* Interactions Modulate the Properties of Cysteine Residues and Disulfide Bonds in Proteins.” Journal of the American Chemical Society 140 (2018): 17606-17611 © 2018 The Author(s) As Published https://dx.doi.org/10.1021/JACS.8B09701 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link https://hdl.handle.net/1721.1/125597 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author J Am Chem Manuscript Author Soc. Author Manuscript Author manuscript; available in PMC 2019 May 21. Published in final edited form as: J Am Chem Soc. 2018 December 19; 140(50): 17606–17611. doi:10.1021/jacs.8b09701. n➝π* Interactions Modulate the Properties of Cysteine Residues and Disulfide Bonds in Proteins Henry R. Kilgore and Ronald T. Raines* Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States Abstract Noncovalent interactions are ubiquitous in biology, taking on roles that include stabilizing the conformation of and assembling biomolecules, and providing an optimal environment for enzymatic catalysis. Here, we describe a noncovalent interaction that engages the sulfur atoms of cysteine residues and disulfide bonds in proteins—their donation of electron density into an antibonding orbital of proximal amide carbonyl groups. -

Validation of a Methodology to Determine Benzene, Toluene
M. L. Gallego-Diez et al.; Revista Facultad de Ingeniería, No. 79, pp. 138-149, 2016 Revista Facultad de Ingeniería, Universidad de Antioquia, No. 79, pp. 138-149, 2016 Validation of a methodology to determine Benzene, Toluene, Ethylbenzene, and Xylenes concentration present in the air and adsorbed in activated charcoal passive samplers by GC/ FID chromatography Validación de una metodología para la determinación de la concentración de Benceno, Tolueno, Etilbenceno y Xilenos, presentes en muestras aire y adsorbidos en captadores pasivos de carbón activado, mediante cromatografía GC/FID Mary Luz Gallego-Díez1*, Mauricio Andrés Correa-Ochoa2, Julio César Saldarriaga-Molina2 1Facultad de Ingeniería, Universidad de Antioquia. Calle 67 # 53- 108. A. A. 1226. Medellín, Colombia. 2Grupo de Ingeniería y Gestión Ambiental (GIGA), Facultad de Ingeniería, Universidad de Antioquia. Calle 67 # 53- 108. A. A. 1226. Medellín, Colombia. ARTICLE INFO ABSTRACT: This article shows the validation of the analytical procedure which allows Received August 28, 2015 determining concentrations of Benzene (B), Toluene (T), Ethylbenzene (E), and Xylenes (X) Accepted April 02, 2016 -compounds known as BTEX- present in the air and adsorbed by over activated charcoal by GC-FID using the (Fluorobenzene) internal standard addition as quantification method. In the process, reference activated charcoal was employed for validation and coconut -base granular charcoal (CGC) for the construction of passive captors used in sample taken in external places or in environmental air. CGC material was selected from its recovering capacity of BTEX, with an average of 89.1% for all analytes. In this research, BTEX presence KEYWORDS in air samples, taken in a road of six lines and characterized for having heavy traffic, in Validation, activated Medellín city (Antioquia, Colombia), was analyzed. -

Technical Notes
1624 Bioconjugate Chem. 2005, 16, 1624−1628 TECHNICAL NOTES Synthesis of Chemical Probes to Map Sulfenic Acid Modifications on Proteins Leslie B. Poole,†* Bu-Bing Zeng,‡,| Sarah A. Knaggs,‡ Mamudu Yakubu,§ and S. Bruce King‡,* Departments of Chemistry and Biochemistry, Wake Forest University, Winston-Salem, North Carolina 27109, and Department of Chemistry, Elizabeth City State University, Elizabeth City, North Carolina 27909 . Received August 23, 2005 Cysteine sulfenic acids in proteins can be identified by their ability to form adducts with dimedone, but this reagent imparts no spectral or affinity tag for subsequent analyses of such tagged proteins. Given its similar reactivity toward cysteine sulfenic acids, 1,3-cyclohexadione was synthetically modified to an alcohol derivative and linked to fluorophores based on isatoic acid and 7-methoxycou- marin. The resulting compounds retain full reactivity and specificity toward cysteine sulfenic acids in proteins, allowing for incorporation of the fluorescent label into the protein and “tagging” it based on its sulfenic acid redox state. Control experiments using dimedone further show the specificity of the reaction of 1,3-diones with protein sulfenic acids in aqueous media. These new compounds provide the basis for an improved method for the detection of protein sulfenic acids. INTRODUCTION Scheme 1 Interest in the identification of cysteine sulfenic acids in proteins by biochemists has grown substantially over the past decade as their biological roles in redox regula- tion and catalysis within an array of cellular proteins have become better defined (1, 2). Despite their impor- tance, only a limited set of tools to identify these species exist, and most of these are only applicable to in vitro studies of pure, isolated proteins (3, 4). -

THIOL OXIDATION a Slippery Slope the Oxidation of Thiols — Molecules RSH Oxidation May Proceed Too Predominates
RESEARCH HIGHLIGHTS Nature Reviews Chemistry | Published online 25 Jan 2017; doi:10.1038/s41570-016-0013 THIOL OXIDATION A slippery slope The oxidation of thiols — molecules RSH oxidation may proceed too predominates. Here, the maximum of the form RSH — can afford quickly for intermediates like RSOH rate constants indicate the order − − − These are many products. From least to most to be spotted and may also afford of reactivity: RSO > RS >> RSO2 . common oxidized, these include disulfides intractable mixtures. Addressing When the reactions are carried out (RSSR), as well as sulfenic (RSOH), the first problem, Chauvin and Pratt in methanol-d , the obtained kinetic reactions, 1 sulfinic (RSO2H) and sulfonic slowed the reactions down by using isotope effect values (kH/kD) are all but have (RSO3H) acids. Such chemistry “very sterically bulky thiols, whose in the range 1.1–1.2, indicating that historically is pervasive in nature, in which corresponding sulfenic acids were no acidic proton is transferred in the been very disulfide bonds between cysteine known to be isolable but were yet rate-determining step. Rather, the residues stabilize protein structures, to be thoroughly explored in terms oxidations involve a specific base- difficult to and where thiols and thiolates often of reactivity”. The second problem catalysed mechanism wherein an study undergo oxidation by H2O2 or O2 in was tackled by modifying the model acid–base equilibrium precedes the order to protect important biological system, 9-mercaptotriptycene, by rate-determining nucleophilic attack − − − structures from damage. Among including a fluorine substituent to of RS , RSO or RSO2 on H2O2. the oxidation products, sulfenic serve as a spectroscopic handle. -

Cysteine Sulfenic Acid As an Intermediate in Disulfide Bond Formation and Nonenzymatic Protein Folding† Douglas S
7748 Biochemistry 2010, 49, 7748–7755 DOI: 10.1021/bi1008694 Cysteine sulfenic Acid as an Intermediate in Disulfide Bond Formation and Nonenzymatic Protein Folding† Douglas S. Rehder and Chad R. Borges* Molecular Biomarkers, The Biodesign Institute at Arizona State University, Tempe, Arizona 85287 Received May 28, 2010; Revised Manuscript Received July 22, 2010 ABSTRACT: As a posttranslational protein modification, cysteine sulfenic acid (Cys-SOH) is well established as an oxidative stress-induced mediator of enzyme function and redox signaling. Data presented herein show that protein Cys-SOH forms spontaneously in air-exposed aqueous solutions of unfolded (disulfide-reduced) protein in the absence of added oxidizing reagents, mediating the oxidative disulfide bond formation process key to in vitro, nonenzymatic protein folding. Molecular oxygen (O2) and trace metals [e.g., copper(II)] are shown to be important reagents in the oxidative refolding process. Cys-SOH is also shown to play a role in spontaneous disulfide-based dimerization of peptide molecules containing free cysteine residues. In total, the data presented expose a chemically ubiquitous role for Cys-SOH in solutions of free cysteine-containing protein exposed to air. In the early 1960s, Anfinsen and colleagues conducted seminal ous oxidants such as hydrogen peroxide are required to build up studies in protein disulfide bond formation and folding, showing significant quantities of Cys-SOH within fully folded proteins. that unfolded (disulfide-reduced) proteins will spontaneously In general, Cys-SOH is a transient intermediate that until the and completely refold, regenerating full protein activity, over a past decade has been difficult to study. It has been understood period of approximately a day in the absence of denaturants that Cys-SOH likely renders the sulfur atom of cysteine electro- (1-6). -

Copper–Sulfenate Complex from Oxidation of a Cavity Mutant of Pseudomonas Aeruginosa Azurin
Copper–sulfenate complex from oxidation of a cavity mutant of Pseudomonas aeruginosa azurin Nathan A. Sierackia,1, Shiliang Tiana,1, Ryan G. Hadtb,1, Jun-Long Zhangc, Julia S. Woertinkb, Mark J. Nilgesa, Furong Suna, Edward I. Solomonb,2, and Yi Lua,2 aDepartment of Chemistry, University of Illinois at Urbana–Champaign, Urbana, IL 61801; bDepartment of Chemistry, Stanford University, Stanford, CA 94305; and cBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, People’s Republic of China Edited by Harry B. Gray, California Institute of Technology, Pasadena, CA, and approved November 27, 2013 (received for review September 3, 2013) Metal–sulfenate centers are known to play important roles in bi- and water. In both enzymes, the sulfenate and sulfinate groups are ology and yet only limited examples are known due to their in- unambiguously required for activity (7, 16). It is commonly postu- stability and high reactivity. Herein we report a copper–sulfenate lated that such oxidized forms of cysteine in a metal site encourage complex characterized in a protein environment, formed at the ac- and stabilize the more oxidized form of redox-active metals, sup- tive site of a cavity mutant of an electron transfer protein, type 1 pressing their potential toxicity in enzymes that do not require the blue copper azurin. Reaction of hydrogen peroxide with Cu(I)– metal center to participate in electron transfer (17, 18). In a more M121G azurin resulted in a species with strong visible absorptions recent example, an iron-coordinated sulfenate was observed when at 350 and 452 nm and a relatively low electron paramagnetic res- a thiolate-containing substrate analog in isopenicillin N-synthase onance gz value of 2.169 in comparison with other normal type 2 was unexpectedly oxidized to a coordinated sulfenato group in copper centers. -
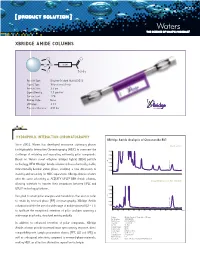
Xbridge Amide Columns
[ PRODUCT SOLUTION ] XBRIDGE AMIDE COLUMNS O O O Si Linker O NH 2 Particle Type: Ethylene Bridged Hybrid [BEH] Ligand Type: Trifunctional Amide Particle Size: 3.5 µm Ligand Density: 7.5 µmol/m2 Carbon Load: 12% Endcap Style: None pH Range: 2-11 Pressure Tolerance: 400 bar HYDROPHILIC INTERacTION CHROMATOGRAPHY XBridge Amide Analysis of Ginsenoside Rb1 Since 2003, Waters has developed innovative stationary phases Orento extract for Hydrophilic Interaction Chromatography [HILIC] to overcome the 0.010 challenge of retaining and separating extremely polar compounds. 0.008 Based on Waters novel ethylene bridged hybrid [BEH] particle 0.006 AU technology, NEW XBridge™ Amide columns utilize a chemically stable, 0.004 0.002 Ginsenoside Rb1 trifunctionally-bonded amide phase, enabling a new dimension in 0.000 stability and versatility for HILIC separations. XBridge Amide columns offer the same selectivity as ACQUITY UPLC® BEH Amide columns, 20 µg/mL Ginsenoside Rb1 standard allowing scientists to transfer their separations between HPLC and 0.010 ® UPLC technology platforms. 0.008 0.006 Designed to retain polar analytes and metabolites that are too polar AU 0.004 to retain by reversed-phase [RP] chromatography, XBridge Amide 0.002 columns facilitate the use of a wide range of mobile phase pH [2 – 11] 0.000 Ginsenoside Rb1 to facilitate the exceptional retention of polar analytes spanning a 0.00. 5 1.01. 5 2.02. 5 3.03. 5 4.04. 5 5.05. 5 6.06. 5 min wide range in polarity, structural moiety and pKa. Column: XBridge Amide, 3.5 µm, 4.6 x 150 mm Part Number: 186004869 Mobile Phase : 80/20 ACN/H2O In addition to enhanced retention of polar compounds, XBridge Flow Rate: 1.4 mL/min Inj. -

Thiolated Polymers: Stability of Thiol Moieties Under Different Storage Conditions
Scientia Pharmaceutica (Sci. Pharm.) 70, 331-339 (2002) 0 Osterreichische Apotheker-Verlagsgesellschaftm.b.H., Wien, Printed in Austria Thiolated polymers: Stability of thiol moieties under different storage conditions A. ~ernko~-schnijrchi*,M.D. ~ornof'*,C.E. ~ast'and N. ~angoth' 'institute of Pharmaceutical Technology and Biopharmaceutics, Center of Pharmacy, University of Vienna, Althanstr. 14, 1090 Vienna, Austria 2~romaPharma GmbH, lndustriezeile 6, 2100 Leobendorf, Austria Abstract: The purpose of this study was to evaluate the stability of thiolated polymers - so-called thiomers. A polycarbophil-cysteine conjugate and a chitosan-thioglycolic acid conjugate were chosen as representative anionic and cationic thiomer. The thiol group bearing compounds L-cysteine and thioglycolic acid were introduced to polycarbophil and chitosan, respectively with a coupling reaction mediated by a carbodiimide. The resulting thiolated polymers were freeze-dried and the amount of thiol groups on the thiomer was determined spectrophotometrically. Each kind of polymer was directly used or compressed into 1 mg matrix-tablets. Polymers were stored for a period of six months at four different storage conditions, namely at -20°C (56% relative humidity; RH), 4°C (53% RH), at 20°C (70% RH), and at 22°C (25% RH). Samples were taken after 6 months to determine the formation of disulfide bonds and the remaining thiol groups on the polymer. When the polycarbophil-cysteine and chitosan-thioglycolic acid conjugate were stored as powder a decrease of free thiol groups was observed only after storage at 20°C and 70% RH. Both polymers were found to be stable under all storage conditions when compressed into matrix tablets.