Flavonoids, Cinnamic Acid Derivatives As Inhibitors of 17(-Hydroxysteroid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

Rosemary)-Derived Ingredients As Used in Cosmetics
Safety Assessment of Rosmarinus Officinalis (Rosemary)-Derived Ingredients as Used in Cosmetics Status: Tentative Amended Report for Public Comment Release Date: March 28, 2014 Panel Meeting Date: June 9-10, 2014 All interested persons are provided 60 days from the above release date to comment on this safety assessment and to identify additional published data that should be included or provide unpublished data which can be made public and included. Information may be submitted without identifying the source or the trade name of the cosmetic product containing the ingredient. All unpublished data submitted to CIR will be discussed in open meetings, will be available at the CIR office for review by any interested party and may be cited in a peer-reviewed scientific journal. Please submit data, comments, or requests to the CIR Director, Dr. Lillian J. Gill. The 2014 Cosmetic Ingredient Review Expert Panel members are: Chairman, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Ronald A. Hill, Ph.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This safety assessment was prepared by Monice M. Fiume, Assistant Director/Senior Scientific Analyst. © Cosmetic Ingredient Review 1620 L Street, NW, Suite 1200♢ Washington, DC 20036 ♢ ph 202.331.0651 ♢ fax 202.331.0088 ♢ [email protected] TABLE OF CONTENTS Abstract ...................................................................................................................................................................................................................................... -

Isocitrate Dehydrogenase 1 (NADP+) (I5036)
Isocitrate Dehydrogenase 1 (NADP+), human recombinant, expressed in Escherichia coli Catalog Number I5036 Storage Temperature –20 °C CAS RN 9028-48-2 IDH1 and IDH2 have frequent genetic alterations in EC 1.1.1.42 acute myeloid leukemia4 and better understanding of Systematic name: Isocitrate:NADP+ oxidoreductase these mutations may lead to an improvement of (decarboxylating) individual cancer risk assessment.6 In addition other studies have shown loss of IDH1 in bladder cancer Synonyms: IDH1, cytosolic NADP(+)-dependent patients during tumor development suggesting this may isocitrate dehydrogenase, isocitrate:NADP+ be involved in tumor progression and metastasis.7 oxidoreductase (decarboxylating), Isocitric Dehydrogenase, ICD1, PICD, IDPC, ICDC, This product is lyophilized from a solution containing oxalosuccinate decarboxylase Tris-HCl, pH 8.0, with trehalose, ammonium sulfate, and DTT. Product Description Isocitrate dehydrogenase (NADP+) [EC 1.1.1.42] is a Purity: ³90% (SDS-PAGE) Krebs cycle enzyme, which converts isocitrate to a-ketoglutarate. The flow of isocitrate through the Specific activity: ³80 units/mg protein glyoxylate bypass is regulated by phosphorylation of isocitrate dehydrogenase, which competes for a Unit definition: 1 unit corresponds to the amount of 1 common substrate (isocitrate) with isocitrate lyase. enzyme, which converts 1 mmole of DL-isocitrate to The activity of the enzyme is dependent on the a-ketoglutarate per minute at pH 7.4 and 37 °C (NADP formation of a magnesium or manganese-isocitrate as cofactor). The activity is measured by observing the 2 complex. reduction of NADP to NADPH at 340 nm in the 7 presence of 4 mM DL-isocitrate and 2 mM MnSO4. -

Pro-Aging Effects of Xanthine Oxidoreductase Products
antioxidants Review Pro-Aging Effects of Xanthine Oxidoreductase Products , , Maria Giulia Battelli y , Massimo Bortolotti y , Andrea Bolognesi * z and Letizia Polito * z Department of Experimental, Diagnostic and Specialty Medicine-DIMES, Alma Mater Studiorum, University of Bologna, Via San Giacomo 14, 40126 Bologna, Italy; [email protected] (M.G.B.); [email protected] (M.B.) * Correspondence: [email protected] (A.B.); [email protected] (L.P.); Tel.: +39-051-20-9-4707 (A.B.); +39-051-20-9-4729 (L.P.) These authors contributed equally. y Co-last authors. z Received: 22 July 2020; Accepted: 4 September 2020; Published: 8 September 2020 Abstract: The senescence process is the result of a series of factors that start from the genetic constitution interacting with epigenetic modifications induced by endogenous and environmental causes and that lead to a progressive deterioration at the cellular and functional levels. One of the main causes of aging is oxidative stress deriving from the imbalance between the production of reactive oxygen (ROS) and nitrogen (RNS) species and their scavenging through antioxidants. Xanthine oxidoreductase (XOR) activities produce uric acid, as well as reactive oxygen and nitrogen species, which all may be relevant to such equilibrium. This review analyzes XOR activity through in vitro experiments, animal studies and clinical reports, which highlight the pro-aging effects of XOR products. However, XOR activity contributes to a regular level of ROS and RNS, which appears essential for the proper functioning of many physiological pathways. This discourages the use of therapies with XOR inhibitors, unless symptomatic hyperuricemia is present. -

RESEARCH COMMUNICATION HADHA Is a Potential Predictor Of
HADHA is a Potential Predictor of the Response to Platinum-based Chemotherapy RESEARCH COMMUNICATION HADHA is a Potential Predictor of Response to Platinum-based Chemotherapy for Lung Cancer Taihei Kageyama1, Ryo Nagashio1, 2, Shinichiro Ryuge 3, Toshihide Matsumoto1,5, Akira Iyoda4, Yukitoshi Satoh4, Noriyuki Masuda3, Shi-Xu Jiang5, Makoto Saegusa5, Yuichi Sato1, 2* Abstract To identify a cisplatin resistance predictor to reduce or prevent unnecessary side effects, we firstly established four cisplatin-resistant sub-lines and compared their protein profiles with cisplatin-sensitive parent lung cancer cell lines using two-dimensional gel electrophoresis. Between the cisplatin-resistant and -sensitive cells, a total of 359 protein spots were differently expressed (>1.5 fold), and 217 proteins (83.0%) were identified. We focused on a mitochondrial protein, hydroxyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase alpha subunit (HADHA), which was increased in all cisplatin-resistant cells. Furthermore, pre- treated biopsy specimens taken from patients who showed resistance to platinum-based treatment showed a significantly higher positive rate for HADHA in all cases (p=0.00367), including non-small cell lung carcinomas (p=0.002), small-cell lung carcinomas (p=0.038), and adenocarcinomas (p=0.008). These results suggest that the expression of HADHA may be a useful marker to predict resistance to platinum-based chemotherapy in patients with lung cancer. Keywords: Cisplatin - HADHA - lung cancer - two-dimensional gel electrophoresis Asian Pacific J Cancer Prev, 12, 3457-3463 Introduction cisplatin resistance rose due to a decrease of blood flow in the tumor and increased DNA repair (Stewart, 2007), Lung cancer is the leading cause of cancer-related the mechanisms underlying cisplatin resistance have not death in the world, and the five-year overall survival rate yet been clarified, and an effective cisplatin resistance is still below 16% (Jemal et al., 2009). -

The Functional Characterization of a Site-Specific Apigenin 4
molecules Article The Functional Characterization of a Site-Specific Apigenin 40-O-methyltransferase Synthesized by the Liverwort Species Plagiochasma appendiculatum Hui Liu, Rui-Xue Xu, Shuai Gao and Ai-Xia Cheng * Key Laboratory of Chemical Biology of Natural Products, Ministry of Education, School of Pharmaceutical Sciences, Shandong University, Jinan 250012, China; [email protected] (H.L.); [email protected] (R.-X.X.); [email protected] (S.G.) * Correspondence: [email protected]; Tel.: +86-531-8838-2012; Fax: +86-531-8838-2019 Academic Editors: Qing-Wen Zhang and Chuangchuang Li Received: 5 April 2017; Accepted: 4 May 2017; Published: 7 May 2017 Abstract: Apigenin, a widely distributed flavone, exhibits excellent antioxidant, anti-inflammatory, and antitumor properties. In addition, the methylation of apigenin is generally considered to result in better absorption and greatly increased bioavailability. Here, four putative Class II methyltransferase genes were identified from the transcriptome sequences generated from the liverwort species Plagiochasma appendiculatum. Each was heterologously expressed as a His-fusion protein in Escherichia coli and their methylation activity against apigenin was tested. One of the four Class II OMT enzymes named 40-O-methyltransferase (Pa40OMT) was shown to react effectively with apigenin, catalyzing its conversion to acacetin. Besides the favorite substrate apigenin, the recombinant PaF40OMT was shown to catalyze luteolin, naringenin, kaempferol, quercetin, genistein, scutellarein, and genkwanin to the corresponding 40-methylation products. In vivo feeding experiments indicated that PaF40OMT could convert apigenin to acacetin efficiently in E. coli and approximately 88.8 µM (25.2 mg/L) of product was synthesized when 100 µM of apigenin was supplemented. -

The Phytochemistry of Cherokee Aromatic Medicinal Plants
medicines Review The Phytochemistry of Cherokee Aromatic Medicinal Plants William N. Setzer 1,2 1 Department of Chemistry, University of Alabama in Huntsville, Huntsville, AL 35899, USA; [email protected]; Tel.: +1-256-824-6519 2 Aromatic Plant Research Center, 230 N 1200 E, Suite 102, Lehi, UT 84043, USA Received: 25 October 2018; Accepted: 8 November 2018; Published: 12 November 2018 Abstract: Background: Native Americans have had a rich ethnobotanical heritage for treating diseases, ailments, and injuries. Cherokee traditional medicine has provided numerous aromatic and medicinal plants that not only were used by the Cherokee people, but were also adopted for use by European settlers in North America. Methods: The aim of this review was to examine the Cherokee ethnobotanical literature and the published phytochemical investigations on Cherokee medicinal plants and to correlate phytochemical constituents with traditional uses and biological activities. Results: Several Cherokee medicinal plants are still in use today as herbal medicines, including, for example, yarrow (Achillea millefolium), black cohosh (Cimicifuga racemosa), American ginseng (Panax quinquefolius), and blue skullcap (Scutellaria lateriflora). This review presents a summary of the traditional uses, phytochemical constituents, and biological activities of Cherokee aromatic and medicinal plants. Conclusions: The list is not complete, however, as there is still much work needed in phytochemical investigation and pharmacological evaluation of many traditional herbal medicines. Keywords: Cherokee; Native American; traditional herbal medicine; chemical constituents; pharmacology 1. Introduction Natural products have been an important source of medicinal agents throughout history and modern medicine continues to rely on traditional knowledge for treatment of human maladies [1]. Traditional medicines such as Traditional Chinese Medicine [2], Ayurvedic [3], and medicinal plants from Latin America [4] have proven to be rich resources of biologically active compounds and potential new drugs. -

Glyoxysomal Malate Dehydrogenase from Watermelon Is Synthesized
Proc. Nati. Acad. Sci. USA Vol. 87, pp. 5773-5777, August 1990 Botany Glyoxysomal malate dehydrogenase from watermelon is synthesized with an amino-terminal transit peptide (isoenzymes/organelle/Citrulus vulgaris/polymerase chain reaction) CHRISTINE GIETL* Institute of Botany, Technical University of Munich, Arcisstrasse 16, D-8000 Munich 2, Federal Republic of Germany; and Department of Physiology, Carlsberg Laboratory, Gamle Carlsberg Vej 10, DK-2500 Copenhagen Valby, Denmark Communicated by Diter von Wettstein, May 11, 1990 (receivedfor review March 5, 1990) ABSTRACT The isolation and sequence of a cDNA clone and peroxisomes are seen between mitochondria and chlo- encoding the complete glyoxysomal malate dehydrogenase roplasts (2). As in plants, mammalian peroxisomes contain [gMDH; (S)-malate:NAD+ oxidoreductase, EC 1.1.1.37] of enzymes involved in the production and degradation ofH202; watermelon cotyledons are presented. Partial cDNA clones in trypanosomes, glycolysis is sequestered into microbodies were synthesized in a three part strategy, taking advantage of called glycosomes (3). All microbodies studied contain en- the polymerase chain reaction technology with oligonucleotides zymes for (3-oxidation and a specific spectrum of other based on directly determined amino acid sequences. Subse- enzymes. It is further characteristic that microbody enzyme quently, the complete done for gMDH was synthesized with a activities are also present in other cell compartments. Gly- sense primer corresponding to the nucleotide sequence of the oxysomes as well as mitochondria contain MDH, citrate N-terminal end of pre-gMDIH and an antisense primer corre- synthase, and enzymes for 83-oxidation (4). The organelle- sponding to the adenylylation site found in the mRNA. -

Cholic Acid for Treating Inborn Errors of Primary Bile Acid Synthesis NHS England Unique Reference Number URN1623 / NICE ID004
NATIONAL INSTITUTE FOR HEALTH AND CARE EXCELLENCE Clinical evidence review of cholic acid for treating inborn errors of primary bile acid synthesis NHS England unique reference number URN1623 / NICE ID004 Prepared by: NICE on behalf of NHS England Specialised Commissioning About this clinical evidence review Clinical evidence reviews are a summary of the best available evidence for a single technology within a licensed indication, for commissioning by NHS England. The clinical evidence review supports NHS England in producing clinical policies but is not NICE guidance or advice. Summary This evidence review considers cholic acid (Laboratoires CTRS [Orphacol] and Retrophin Europe Ltd [Kolbam]) for treating inborn errors of primary bile acid NICE clinical evidence review of cholic acid for treating inborn errors of primary bile acid synthesis Page 1 of 72 NHS URN1623 NICE ID004 synthesis caused by the following enzyme deficiencies in people aged 1 month and over: 3-beta-hydroxy-delta5-C27-steroid oxidoreductase (3beta-HSD) delta4-3-oxosteroid-5-beta reductase (5beta-reductase) 2- (or alpha-) methylacyl-CoA racemase (AMACR) sterol 27-hydroxylase (presenting as cerebrotendinous xanthomatosis [CTX]) cholesterol 7alpha-hydroxylase (CYP7A1). Inborn errors of primary bile acid synthesis are rare genetic conditions in which enzyme deficiencies prevent the liver from converting cholesterol in the body to bile acids (such as cholic acid and chenodeoxycholic acid). This results in the liver producing high concentrations of atypical (or ‘unusual’) bile acids and intermediary metabolites (some of which are toxic to the liver) in an attempt to establish a normal bile acid pool. Accumulation of potentially toxic atypical bile acids and metabolites, and reduced flow of bile acids may cause liver injury. -
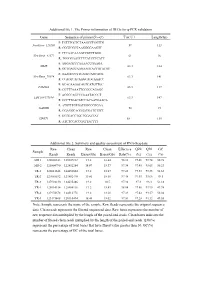
Additional File 1
Additional file 1. The Primer information of DEGs for q-PCR validation Gene Sequence of primer(5'→3') Tm(℃) Length(bp) F: TGTTTGCTCTAAGCCTGGTTG NewGene_126260 59 113 R: CGGTCGCTAAGGGGAAGTT F: CTCAACAAAGCCGTCTGGG NewGene_41572 61 96 R: TGGGGAATCTTCATCCTCATT F: AGGAGCCCAAAACCGAAGA MME 63.3 184 R: GCTGACCAAGAAGTACCGTATGT F: AAAGCCCTTCAGTCAGCACG NewGene_70974 63.3 141 R: CCAGTCACAAGCAGCAAACC F: GCACAAGGCAGTCATGTTGC FAM43A 63.3 117 R: CGTTTAAATTCCGCCAGAGC F: ACGGCAGCCCAAATACCCT LOC108177184 63.3 147 R: GCCTTGACATCCACAATGAACA F: ATGTTTGTGATGGGCGTGAA GAPDH 58 94 R: GGAGGCAGGGATGATGTTCT F: GCTGACCTGCTGGATTAT HPRT1 58 135 R: ATCTCCACCGATTACTTT Additional file 2. Summary and quality assessment of RNA-Seq data Raw Clean Raw Clean Effective Q20 Q30 GC Sample Reads Reads Bases(Gb) Bases(Gb) Rate(%) (%) (%) (%) MR-1 128008342 125607312 19.2 18.84 98.12 97.46 93.74 50.78 MR-2 125804790 122452284 18.87 18.37 97.34 97.45 93.65 50.23 TR-1 128011648 124452604 19.2 18.67 97.22 97.33 93.35 51.65 TR-2 127085532 123882470 19.06 18.58 97.48 97.45 93.66 49.5 TR-3 127994130 124635486 19.2 18.7 97.38 97.3 93.3 51.18 YR-1 128014314 125504116 19.2 18.83 98.04 97.56 93.95 49.98 YR-2 127974678 124512778 19.2 18.68 97.29 97.42 93.57 50.04 YR-3 123173408 120110494 18.48 18.02 97.51 97.28 93.32 49.55 Note: Sample represents the name of the sample. Raw Reads represents the original sequence data. Clean reads represents the filtered sequenced data. Raw bases represents the number of raw sequence data multiplied by the length of the paired-end reads. -

Bioactivity of Curcumin on the Cytochrome P450 Enzymes of the Steroidogenic Pathway
International Journal of Molecular Sciences Article Bioactivity of Curcumin on the Cytochrome P450 Enzymes of the Steroidogenic Pathway Patricia Rodríguez Castaño 1,2, Shaheena Parween 1,2 and Amit V Pandey 1,2,* 1 Pediatric Endocrinology, Diabetology, and Metabolism, University Children’s Hospital Bern, 3010 Bern, Switzerland; [email protected] (P.R.C.); [email protected] (S.P.) 2 Department of Biomedical Research, University of Bern, 3010 Bern, Switzerland * Correspondence: [email protected]; Tel.: +41-31-632-9637 Received: 5 September 2019; Accepted: 16 September 2019; Published: 17 September 2019 Abstract: Turmeric, a popular ingredient in the cuisine of many Asian countries, comes from the roots of the Curcuma longa and is known for its use in Chinese and Ayurvedic medicine. Turmeric is rich in curcuminoids, including curcumin, demethoxycurcumin, and bisdemethoxycurcumin. Curcuminoids have potent wound healing, anti-inflammatory, and anti-carcinogenic activities. While curcuminoids have been studied for many years, not much is known about their effects on steroid metabolism. Since many anti-cancer drugs target enzymes from the steroidogenic pathway, we tested the effect of curcuminoids on cytochrome P450 CYP17A1, CYP21A2, and CYP19A1 enzyme activities. When using 10 µg/mL of curcuminoids, both the 17α-hydroxylase as well as 17,20 lyase activities of CYP17A1 were reduced significantly. On the other hand, only a mild reduction in CYP21A2 activity was observed. Furthermore, CYP19A1 activity was also reduced up to ~20% of control when using 1–100 µg/mL of curcuminoids in a dose-dependent manner. Molecular docking studies confirmed that curcumin could dock onto the active sites of CYP17A1, CYP19A1, as well as CYP21A2. -

Potential Role of Flavonoids in Treating Chronic Inflammatory Diseases with a Special Focus on the Anti-Inflammatory Activity of Apigenin
Review Potential Role of Flavonoids in Treating Chronic Inflammatory Diseases with a Special Focus on the Anti-Inflammatory Activity of Apigenin Rashida Ginwala, Raina Bhavsar, DeGaulle I. Chigbu, Pooja Jain and Zafar K. Khan * Department of Microbiology and Immunology, and Center for Molecular Virology and Neuroimmunology, Center for Cancer Biology, Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine, Philadelphia, PA 19129, USA; [email protected] (R.G.); [email protected] (R.B.); [email protected] (D.I.C.); [email protected] (P.J.) * Correspondence: [email protected] Received: 28 November 2018; Accepted: 30 January 2019; Published: 5 February 2019 Abstract: Inflammation has been reported to be intimately linked to the development or worsening of several non-infectious diseases. A number of chronic conditions such as cancer, diabetes, cardiovascular disorders, autoimmune diseases, and neurodegenerative disorders emerge as a result of tissue injury and genomic changes induced by constant low-grade inflammation in and around the affected tissue or organ. The existing therapies for most of these chronic conditions sometimes leave more debilitating effects than the disease itself, warranting the advent of safer, less toxic, and more cost-effective therapeutic alternatives for the patients. For centuries, flavonoids and their preparations have been used to treat various human illnesses, and their continual use has persevered throughout the ages. This review focuses on the anti-inflammatory actions of flavonoids against chronic illnesses such as cancer, diabetes, cardiovascular diseases, and neuroinflammation with a special focus on apigenin, a relatively less toxic and non-mutagenic flavonoid with remarkable pharmacodynamics. Additionally, inflammation in the central nervous system (CNS) due to diseases such as multiple sclerosis (MS) gives ready access to circulating lymphocytes, monocytes/macrophages, and dendritic cells (DCs), causing edema, further inflammation, and demyelination.