Cholic Acid for Treating Inborn Errors of Primary Bile Acid Synthesis NHS England Unique Reference Number URN1623 / NICE ID004
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

Isocitrate Dehydrogenase 1 (NADP+) (I5036)
Isocitrate Dehydrogenase 1 (NADP+), human recombinant, expressed in Escherichia coli Catalog Number I5036 Storage Temperature –20 °C CAS RN 9028-48-2 IDH1 and IDH2 have frequent genetic alterations in EC 1.1.1.42 acute myeloid leukemia4 and better understanding of Systematic name: Isocitrate:NADP+ oxidoreductase these mutations may lead to an improvement of (decarboxylating) individual cancer risk assessment.6 In addition other studies have shown loss of IDH1 in bladder cancer Synonyms: IDH1, cytosolic NADP(+)-dependent patients during tumor development suggesting this may isocitrate dehydrogenase, isocitrate:NADP+ be involved in tumor progression and metastasis.7 oxidoreductase (decarboxylating), Isocitric Dehydrogenase, ICD1, PICD, IDPC, ICDC, This product is lyophilized from a solution containing oxalosuccinate decarboxylase Tris-HCl, pH 8.0, with trehalose, ammonium sulfate, and DTT. Product Description Isocitrate dehydrogenase (NADP+) [EC 1.1.1.42] is a Purity: ³90% (SDS-PAGE) Krebs cycle enzyme, which converts isocitrate to a-ketoglutarate. The flow of isocitrate through the Specific activity: ³80 units/mg protein glyoxylate bypass is regulated by phosphorylation of isocitrate dehydrogenase, which competes for a Unit definition: 1 unit corresponds to the amount of 1 common substrate (isocitrate) with isocitrate lyase. enzyme, which converts 1 mmole of DL-isocitrate to The activity of the enzyme is dependent on the a-ketoglutarate per minute at pH 7.4 and 37 °C (NADP formation of a magnesium or manganese-isocitrate as cofactor). The activity is measured by observing the 2 complex. reduction of NADP to NADPH at 340 nm in the 7 presence of 4 mM DL-isocitrate and 2 mM MnSO4. -

Pro-Aging Effects of Xanthine Oxidoreductase Products
antioxidants Review Pro-Aging Effects of Xanthine Oxidoreductase Products , , Maria Giulia Battelli y , Massimo Bortolotti y , Andrea Bolognesi * z and Letizia Polito * z Department of Experimental, Diagnostic and Specialty Medicine-DIMES, Alma Mater Studiorum, University of Bologna, Via San Giacomo 14, 40126 Bologna, Italy; [email protected] (M.G.B.); [email protected] (M.B.) * Correspondence: [email protected] (A.B.); [email protected] (L.P.); Tel.: +39-051-20-9-4707 (A.B.); +39-051-20-9-4729 (L.P.) These authors contributed equally. y Co-last authors. z Received: 22 July 2020; Accepted: 4 September 2020; Published: 8 September 2020 Abstract: The senescence process is the result of a series of factors that start from the genetic constitution interacting with epigenetic modifications induced by endogenous and environmental causes and that lead to a progressive deterioration at the cellular and functional levels. One of the main causes of aging is oxidative stress deriving from the imbalance between the production of reactive oxygen (ROS) and nitrogen (RNS) species and their scavenging through antioxidants. Xanthine oxidoreductase (XOR) activities produce uric acid, as well as reactive oxygen and nitrogen species, which all may be relevant to such equilibrium. This review analyzes XOR activity through in vitro experiments, animal studies and clinical reports, which highlight the pro-aging effects of XOR products. However, XOR activity contributes to a regular level of ROS and RNS, which appears essential for the proper functioning of many physiological pathways. This discourages the use of therapies with XOR inhibitors, unless symptomatic hyperuricemia is present. -

RESEARCH COMMUNICATION HADHA Is a Potential Predictor Of
HADHA is a Potential Predictor of the Response to Platinum-based Chemotherapy RESEARCH COMMUNICATION HADHA is a Potential Predictor of Response to Platinum-based Chemotherapy for Lung Cancer Taihei Kageyama1, Ryo Nagashio1, 2, Shinichiro Ryuge 3, Toshihide Matsumoto1,5, Akira Iyoda4, Yukitoshi Satoh4, Noriyuki Masuda3, Shi-Xu Jiang5, Makoto Saegusa5, Yuichi Sato1, 2* Abstract To identify a cisplatin resistance predictor to reduce or prevent unnecessary side effects, we firstly established four cisplatin-resistant sub-lines and compared their protein profiles with cisplatin-sensitive parent lung cancer cell lines using two-dimensional gel electrophoresis. Between the cisplatin-resistant and -sensitive cells, a total of 359 protein spots were differently expressed (>1.5 fold), and 217 proteins (83.0%) were identified. We focused on a mitochondrial protein, hydroxyl-coenzyme A dehydrogenase/3-ketoacyl-coenzyme A thiolase/enoyl-coenzyme A hydratase alpha subunit (HADHA), which was increased in all cisplatin-resistant cells. Furthermore, pre- treated biopsy specimens taken from patients who showed resistance to platinum-based treatment showed a significantly higher positive rate for HADHA in all cases (p=0.00367), including non-small cell lung carcinomas (p=0.002), small-cell lung carcinomas (p=0.038), and adenocarcinomas (p=0.008). These results suggest that the expression of HADHA may be a useful marker to predict resistance to platinum-based chemotherapy in patients with lung cancer. Keywords: Cisplatin - HADHA - lung cancer - two-dimensional gel electrophoresis Asian Pacific J Cancer Prev, 12, 3457-3463 Introduction cisplatin resistance rose due to a decrease of blood flow in the tumor and increased DNA repair (Stewart, 2007), Lung cancer is the leading cause of cancer-related the mechanisms underlying cisplatin resistance have not death in the world, and the five-year overall survival rate yet been clarified, and an effective cisplatin resistance is still below 16% (Jemal et al., 2009). -

|||||||||||||III US005202354A United States Patent (19) (11) Patent Number: 5,202,354 Matsuoka Et Al
|||||||||||||III US005202354A United States Patent (19) (11) Patent Number: 5,202,354 Matsuoka et al. 45) Date of Patent: Apr. 13, 1993 (54) COMPOSITION AND METHOD FOR 4,528,295 7/1985 Tabakoff .......... ... 514/562 X REDUCING ACETALDEHYDE TOXCTY 4,593,020 6/1986 Guinot ................................ 514/811 (75) Inventors: Masayoshi Matsuoka, Habikino; Go OTHER PUBLICATIONS Kito, Yao, both of Japan Sprince et al., Agents and Actions, vol. 5/2 (1975), pp. 73) Assignee: Takeda Chemical Industries, Ltd., 164-173. Osaka, Japan Primary Examiner-Arthur C. Prescott (21) Appl. No.: 839,265 Attorney, Agent, or Firn-Wenderoth, Lind & Ponack 22) Filed: Feb. 21, 1992 (57) ABSTRACT A novel composition and method are disclosed for re Related U.S. Application Data ducing acetaldehyde toxicity, especially for preventing (63) Continuation of Ser. No. 13,443, Feb. 10, 1987, aban and relieving hangover symptoms in humans. The com doned. position comprises (a) a compound of the formula: (30) Foreign Application Priority Data Feb. 18, 1986 JP Japan .................................. 61-34494 51) Int: C.5 ..................... A01N 37/00; A01N 43/08 52 U.S. C. ................................. ... 514/562; 514/474; 514/81 wherein R is hydrogen or an acyl group; R' is thiol or 58) Field of Search ................ 514/557, 562, 474,811 sulfonic group; and n is an integer of 1 or 2, (b) ascorbic (56) References Cited acid or a salt thereof and (c) a disulfide type thiamine derivative or a salt thereof. The composition is orally U.S. PATENT DOCUMENTS administered, preferably in the form of tablets. 2,283,817 5/1942 Martin et al. -

OCALIVA™ (Obeticholic Acid) Oral
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 9/15/2016 SECTION: DRUGS LAST REVIEW DATE: 8/19/2021 LAST CRITERIA REVISION DATE: 8/19/2021 ARCHIVE DATE: OCALIVA™ (obeticholic acid) oral Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. BLUE CROSS®, BLUE SHIELD® and the Cross and Shield Symbols are registered service marks of the Blue Cross and Blue Shield Association, an association of independent Blue Cross and Blue Shield Plans. -

Glyoxysomal Malate Dehydrogenase from Watermelon Is Synthesized
Proc. Nati. Acad. Sci. USA Vol. 87, pp. 5773-5777, August 1990 Botany Glyoxysomal malate dehydrogenase from watermelon is synthesized with an amino-terminal transit peptide (isoenzymes/organelle/Citrulus vulgaris/polymerase chain reaction) CHRISTINE GIETL* Institute of Botany, Technical University of Munich, Arcisstrasse 16, D-8000 Munich 2, Federal Republic of Germany; and Department of Physiology, Carlsberg Laboratory, Gamle Carlsberg Vej 10, DK-2500 Copenhagen Valby, Denmark Communicated by Diter von Wettstein, May 11, 1990 (receivedfor review March 5, 1990) ABSTRACT The isolation and sequence of a cDNA clone and peroxisomes are seen between mitochondria and chlo- encoding the complete glyoxysomal malate dehydrogenase roplasts (2). As in plants, mammalian peroxisomes contain [gMDH; (S)-malate:NAD+ oxidoreductase, EC 1.1.1.37] of enzymes involved in the production and degradation ofH202; watermelon cotyledons are presented. Partial cDNA clones in trypanosomes, glycolysis is sequestered into microbodies were synthesized in a three part strategy, taking advantage of called glycosomes (3). All microbodies studied contain en- the polymerase chain reaction technology with oligonucleotides zymes for (3-oxidation and a specific spectrum of other based on directly determined amino acid sequences. Subse- enzymes. It is further characteristic that microbody enzyme quently, the complete done for gMDH was synthesized with a activities are also present in other cell compartments. Gly- sense primer corresponding to the nucleotide sequence of the oxysomes as well as mitochondria contain MDH, citrate N-terminal end of pre-gMDIH and an antisense primer corre- synthase, and enzymes for 83-oxidation (4). The organelle- sponding to the adenylylation site found in the mRNA. -

Ocaliva (Obeticholic Acid)
HIGHLIGHTS OF PRESCRIBING INFORMATION Staging/ Classification Non-Cirrhotic or Child-Pugh Class B These highlights do not include all the information needed to use Compensated Child- or C or Patients OCALIVA® safely and effectively. See full prescribing information for Pugh Class A with a Prior OCALIVA. Decompensation a Event ® OCALIVA (obeticholic acid) tablets, for oral use Starting OCALIVA 5 mg once daily 5 mg once weekly Initial U.S. Approval: 2016 Dosage for first 3 months WARNING: HEPATIC DECOMPENSATION AND FAILURE IN OCALIVA Dosage 10 mg once daily 5 mg twice weekly INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH Titration after first (at least 3 days apart) CLASS B OR C OR DECOMPENSATED CIRRHOSIS 3 months, for patients See full prescribing information for complete boxed warning who have not achieved Titrate to 10 mg an adequate reduction twice weekly (at least • In postmarketing reports, hepatic decompensation and failure, in in ALP and/or total 3 days apart) based bilirubin and who are on response and some cases fatal, have been reported in patients with primary biliary b tolerating OCALIVA tolerability cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more Maximum OCALIVA 10 mg once daily 10 mg twice weekly frequently than recommended. (5.1) Dosage (at least 3 days apart) a • The recommended starting dosage of OCALIVA is 5 mg once weekly Gastroesophageal variceal bleeding, new or worsening jaundice, for patients with Child-Pugh Class B or C hepatic impairment or a spontaneous bacterial peritonitis, etc. b prior decompensation event. -

Biliary Bile Acid Composition of the Human Fetus in Early Gestation
0031-3998/87/2102-0197$02.00/0 PEDIATRIC RESEARCH Vol. 21, No.2, 1987 Copyright© 1987 International Pediatric Research Foundation, Inc. Printed in U.S.A. Biliary Bile Acid Composition of the Human Fetus in Early Gestation C. COLOMBO, G. ZULIANI, M. RONCHI, J. BREIDENSTEIN, AND K. D. R. SETCHELL Department q/Pediatrics and Obstetrics, University q/ Milan, Milan, Italy [C. C., G.Z., M.R.j and Department q/ Pediatric Gastroel1/erology and Nwrition, Children's Hospital Medical Center, Cincinnati, Ohio 45229 [J.B., K.D.R.S.j ABSTRACf. Using analytical techniques, which included the key processes of the enterohepatic circulation of bile acids capillary column gas-liquid chromatography and mass ( 16-19). As a result of physiologic cholestasis, the newborn infant spectrometry, detailed bile acid profiles were obtained for has a tendency to develop a true neonatal cholestasis when 24 fetal bile samples collected after legal abortions were subjected to such stresses as sepsis, hypoxia, and total parenteral performed between the 14th and 20th wk of gestation. nutrition ( 15). Qualitatively, the bile acid profiles of all fetal bile samples Abnormalities in bile acid metabolism have been suggested to were similar. The predominant bile acids identified were be a factor in the development of certain forms of cholestasis in chenodeoxycholic and cholic acid. The presence of small the newborn (20) and in animal models the hepatotoxic nature but variable amounts of deoxycholic acid and traces of of bile acids has been well established (21-24). A greater under lithocholic acid suggested placental transfer of these bile standing of the etiology of these conditions requires a better acids from the maternal circulation. -
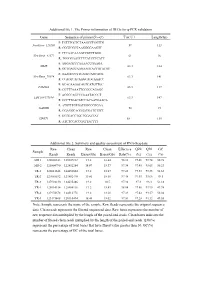
Additional File 1
Additional file 1. The Primer information of DEGs for q-PCR validation Gene Sequence of primer(5'→3') Tm(℃) Length(bp) F: TGTTTGCTCTAAGCCTGGTTG NewGene_126260 59 113 R: CGGTCGCTAAGGGGAAGTT F: CTCAACAAAGCCGTCTGGG NewGene_41572 61 96 R: TGGGGAATCTTCATCCTCATT F: AGGAGCCCAAAACCGAAGA MME 63.3 184 R: GCTGACCAAGAAGTACCGTATGT F: AAAGCCCTTCAGTCAGCACG NewGene_70974 63.3 141 R: CCAGTCACAAGCAGCAAACC F: GCACAAGGCAGTCATGTTGC FAM43A 63.3 117 R: CGTTTAAATTCCGCCAGAGC F: ACGGCAGCCCAAATACCCT LOC108177184 63.3 147 R: GCCTTGACATCCACAATGAACA F: ATGTTTGTGATGGGCGTGAA GAPDH 58 94 R: GGAGGCAGGGATGATGTTCT F: GCTGACCTGCTGGATTAT HPRT1 58 135 R: ATCTCCACCGATTACTTT Additional file 2. Summary and quality assessment of RNA-Seq data Raw Clean Raw Clean Effective Q20 Q30 GC Sample Reads Reads Bases(Gb) Bases(Gb) Rate(%) (%) (%) (%) MR-1 128008342 125607312 19.2 18.84 98.12 97.46 93.74 50.78 MR-2 125804790 122452284 18.87 18.37 97.34 97.45 93.65 50.23 TR-1 128011648 124452604 19.2 18.67 97.22 97.33 93.35 51.65 TR-2 127085532 123882470 19.06 18.58 97.48 97.45 93.66 49.5 TR-3 127994130 124635486 19.2 18.7 97.38 97.3 93.3 51.18 YR-1 128014314 125504116 19.2 18.83 98.04 97.56 93.95 49.98 YR-2 127974678 124512778 19.2 18.68 97.29 97.42 93.57 50.04 YR-3 123173408 120110494 18.48 18.02 97.51 97.28 93.32 49.55 Note: Sample represents the name of the sample. Raw Reads represents the original sequence data. Clean reads represents the filtered sequenced data. Raw bases represents the number of raw sequence data multiplied by the length of the paired-end reads. -

G Protein-Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. British Journal of Pharmacology (2015) 172, 5744–5869 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: G protein-coupled receptors Stephen PH Alexander1, Anthony P Davenport2, Eamonn Kelly3, Neil Marrion3, John A Peters4, Helen E Benson5, Elena Faccenda5, Adam J Pawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Biomedical Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK, 2Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK, 3School of Physiology and Pharmacology, University of Bristol, Bristol, BS8 1TD, UK, 4Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK, 5Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2015/16 provides concise overviews of the key properties of over 1750 human drug targets with their pharmacology, plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. The full contents can be found at http://onlinelibrary.wiley.com/doi/ 10.1111/bph.13348/full. G protein-coupled receptors are one of the eight major pharmacological targets into which the Guide is divided, with the others being: ligand-gated ion channels, voltage-gated ion channels, other ion channels, nuclear hormone receptors, catalytic receptors, enzymes and transporters. These are presented with nomenclature guidance and summary information on the best available pharmacological tools, alongside key references and suggestions for further reading. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01