Tip-Enhanced Laser Ablation Sample Transfer for Mass Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Daily Iowan (Iowa City, Iowa), 1953-10-22
The Weather Serving the State Partb c10., aDd eeoIH ·versity of Iowa toda,. CleariDc ....... Art slderlbb eooIer FrWar. Campus and JII,h ~aJ. T.; 1_. 52; Hla'h WednH41a7. II; low. he SUI art i.:wa City 67. attend the 1868 - A,P Leased Wile, Wirephot.o - Five Cent. Iowa City, Iowa, Thursday, Oct. 22, 1953 - Vol. 97, No. 276 the found- , Eat. College Art • ~---------------------------------------------- Department orlSin. Mln ./ '0 the rti.t trom .ohio the conter- ~ shown by ueen ·Will· Open Homecoming ence df 1he AcsthetJcs ege in. No- • Homecoming Mums Aqui Show To Feature Five Iowa sportscasters and Qne ------------ Varied Acts sports writer will judge the utes past any given point In the beauty, or~inallty and humor ot parade route. The 40 tloats wiU Three days ot 1953 Homecom , ' the I10nt entries In the fifth an- then move to the area just east In, frolic will let the ful\-steam- nual SUI Homecoming parade Frl- of Iowa stadium to remain throu,ll abead si,nal tonight when one ot day night at 7. Saturday's game as a post-parade Taking time out from their re- dISplay parade chairman Loren five campus lovelies is named porting responsibilities to score Hlcker ~on lays. Dolphin Queen for SUY'! 31st an- each of 40 floats ror beaut), will . nUIII poolsld~ sholl'. be Gene Claussen, KXIC Iowa Musical marching grou~s In the City and Gene Shumate KSO De parade will total 13, With nine Pre!\Cntatlon of the qucen-ror- Moines. • high school bands, Shriner bands a-weekend will come at 8 p.m. -

Quantitative Force Measurements with Optical Tweezers: the JPK Nanotracker™
Quantitative force measurements with optical tweezers: The JPK NanoTracker™ Introduction to optical tweezers manipulate biomolecules and cells, but also to directly and accurately measure the minute forces (on the order of Optical tweezers methodology has developed from proof-of- fractions of picoNewtons) involved. Most often, the principle experiments to an established quantitative biomolecules of interest are not trapped themselves technique in fields ranging from (bio)physics through cell directly, but manipulated through functionalized biology. As the name suggests, optical tweezers are a microspheres. means to manipulate objects with light. With this technique, The correct physical description of optical trapping depends microscopically small objects can be held and manipulated. on the size of the trapped object. One speaks of the ‘ray- At the same time, the forces exerted on the trapped objects optics’ regime when the object’s dimension d is much larger can be accurately measured. The basic physical principle than the wavelength of the trapping light: d>>λ. In this case, underlying optical tweezers is the radiation pressure diffraction effects can be neglected and the trapping forces exerted by light when colliding with matter. For macroscopic of the light can be understood in terms of ray optics. The objects, the radiation pressure exerted by typical light regime where d<<λ is called the Rayleigh regime. In this sources is orders of magnitude too small to have any case, the trapped particles can be treated as point dipoles, measurable effect: we do not feel the light power of the sun as the electromagnetic field is constant on the scale of the pushing us away. -
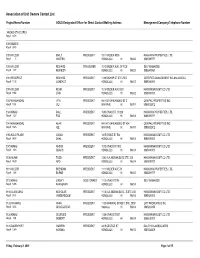
Association of Unit Owners Contact List
Association of Unit Owners Contact List Project Name/Number AOUO Designated Officer for Direct Contact/Mailing Address Management Company/Telephone Number `AKOKO AT HO`OPILI Reg.# 8073 1001 QUEEN Reg.# 7675 1001 WILDER EMILY PRESIDENT 1001 WILDER #305 HAWAIIAN PROPERTIES, LTD. Reg.# 5 WATERS HONOLULU HI 96822 8085399777 1010 WILDER RICHARD TREASURER 1010 WILDER AVE, OFFICE SELF MANAGED Reg.# 377 KENNEDY HONOLULU HI 96822 8085241961 1011 PROSPECT RICHARD PRESIDENT 1188 BISHOP ST STE 2503 CERTIFIED MANAGEMENT INC dba ASSOCI Reg.# 1130 CONRADT HONOLULU HI 96813 8088360911 1015 WILDER KEVIN PRESIDENT 1015 WILDER AVE #201 HAWAIIANA MGMT CO LTD Reg.# 1960 LIMA HONOLULU HI 96822 8085939100 1037 KAHUAMOKU VITA PRESIDENT 94-1037 KAHUAMOKU ST 3 CEN PAC PROPERTIES INC Reg.# 1551 VILI WAIPAHU HI 96797 8085932902 1040 KINAU PAUL PRESIDENT 1040 KINAU ST., #1206 HAWAIIAN PROPERTIES, LTD. Reg.# 527 FOX HONOLULU HI 96814 8085399777 1041 KAHUAMOKU ALAN PRESIDENT 94-1041 KAHUAMOKU ST 404 CEN PAC PROPERTIES INC Reg.# 1623 IGE WAIPAHU HI 96797 8085932902 1054 KALO PLACE JUANA PRESIDENT 1415 S KING ST 504 HAWAIIANA MGMT CO LTD Reg.# 5450 DAHL HONOLULU HI 96814 8085939100 1073 KINAU ANSON PRESIDENT 1073 KINAU ST 1003 HAWAIIANA MGMT CO LTD Reg.# 616 QUACH HONOLULU HI 96814 8085939100 1108 AUAHI TODD PRESIDENT 1240 ALA MOANA BLVD STE. 200 HAWAIIANA MGMT CO LTD Reg.# 7429 APO HONOLULU HI 96814 8085939100 1111 WILDER BRENDAN PRESIDENT 1111 WILDER AVE 7A HAWAIIAN PROPERTIES, LTD. Reg.# 228 BURNS HONOLULU HI 96822 8085399777 1112 KINAU LINDA Y SOLE OWNER 1112 KINAU ST PH SELF MANAGED Reg.# 1295 NAKAGAWA HONOLULU HI 96814 1118 ALA MOANA NICHOLAS PRESIDENT 1118 ALA MOANA BLVD., SUITE 200 HAWAIIANA MGMT CO LTD Reg.# 7431 VANDERBOOM HONOLULU HI 96814 8085939100 1133 WAIMANU ANNA PRESIDENT 1133 WAIMANU STREET, STE. -

A Quantum Origin of Life?
June 26, 2008 11:1 World Scientific Book - 9in x 6in quantum Chapter 1 A Quantum Origin of Life? Paul C. W. Davies The origin of life is one of the great unsolved problems of science. In the nineteenth century, many scientists believed that life was some sort of magic matter. The continued use of the term “organic chemistry” is a hangover from that era. The assumption that there is a chemical recipe for life led to the hope that, if only we knew the details, we could mix up the right stuff in a test tube and make life in the lab. Most research on biogenesis has followed that tradition, by assuming chemistry was a bridge—albeit a long one—from matter to life. Elucidat- ing the chemical pathway has been a tantalizing goal, spurred on by the famous Miller-Urey experiment of 1952, in which amino acids were made by sparking electricity through a mixture of water and common gases [Miller (1953)]. But this concept turned out to be something of a blind alley, and further progress with pre-biotic chemical synthesis has been frustratingly slow. In 1944, Erwin Schr¨odinger published his famous lectures under the ti- tle What is Life? [Schr¨odinger (1944)] and ushered in the age of molecular biology. Sch¨odinger argued that the stable transmission of genetic infor- mation from generation to generation in discrete bits implied a quantum mechanical process, although he was unaware of the role of or the specifics of genetic encoding. The other founders of quantum mechanics, including Niels Bohr, Werner Heisenberg and Eugene Wigner shared Schr¨odinger’s belief that quantum physics was the key to understanding the phenomenon Received May 9, 2007 3 QUANTUM ASPECTS OF LIFE © Imperial College Press http://www.worldscibooks.com/physics/p581.html June 26, 2008 11:1 World Scientific Book - 9in x 6in quantum 4 Quantum Aspects of Life of life. -

Martian Crater Morphology
ANALYSIS OF THE DEPTH-DIAMETER RELATIONSHIP OF MARTIAN CRATERS A Capstone Experience Thesis Presented by Jared Howenstine Completion Date: May 2006 Approved By: Professor M. Darby Dyar, Astronomy Professor Christopher Condit, Geology Professor Judith Young, Astronomy Abstract Title: Analysis of the Depth-Diameter Relationship of Martian Craters Author: Jared Howenstine, Astronomy Approved By: Judith Young, Astronomy Approved By: M. Darby Dyar, Astronomy Approved By: Christopher Condit, Geology CE Type: Departmental Honors Project Using a gridded version of maritan topography with the computer program Gridview, this project studied the depth-diameter relationship of martian impact craters. The work encompasses 361 profiles of impacts with diameters larger than 15 kilometers and is a continuation of work that was started at the Lunar and Planetary Institute in Houston, Texas under the guidance of Dr. Walter S. Keifer. Using the most ‘pristine,’ or deepest craters in the data a depth-diameter relationship was determined: d = 0.610D 0.327 , where d is the depth of the crater and D is the diameter of the crater, both in kilometers. This relationship can then be used to estimate the theoretical depth of any impact radius, and therefore can be used to estimate the pristine shape of the crater. With a depth-diameter ratio for a particular crater, the measured depth can then be compared to this theoretical value and an estimate of the amount of material within the crater, or fill, can then be calculated. The data includes 140 named impact craters, 3 basins, and 218 other impacts. The named data encompasses all named impact structures of greater than 100 kilometers in diameter. -

Depth-Resolved Measurement of Optical Radiation-Pressure Forces with Optical Coherence Tomography
Vol. 26, No. 3 | 5 Feb 2018 | OPTICS EXPRESS 2410 Depth-resolved measurement of optical radiation-pressure forces with optical coherence tomography NICHALUK LEARTPRAPUN, RISHYASHRING R. IYER, AND STEVEN G. ADIE* Meinig School of Biomedical Engineering, Cornell University, Ithaca, NY 14853, USA *[email protected] Abstract: A weakly focused laser beam can exert sufficient radiation pressure to manipulate microscopic particles over a large depth range. However, depth-resolved continuous measurement of radiation-pressure force profiles over an extended range about the focal plane has not been demonstrated despite decades of research on optical manipulation. Here, we present a method for continuous measurement of axial radiation-pressure forces from a weakly focused beam on polystyrene micro-beads suspended in viscous fluids over a depth range of 400 μm, based on real-time monitoring of particle dynamics using optical coherence tomography (OCT). Measurements of radiation-pressure forces as a function of beam power, wavelength, bead size, and refractive index are consistent with theoretical trends. However, our continuous measurements also reveal localized depth-dependent features in the radiation- pressure force profiles that deviate from theoretical predictions based on an aberration-free Gaussian beam. The combination of long-range radiation pressure and OCT offers a new mode of quantitative optical manipulation and detection with extended spatial coverage. This may find applications in the characterization of optical tractor beams, or volumetric optical manipulation and interrogation of beads in viscoelastic media. © 2018 Optical Society of America under the terms of the OSA Open Access Publishing Agreement OCIS codes: (110.4500) Optical coherence tomography; (350.4855) Optical tweezers or optical manipulation. -

Planetary Constants and Models
JPL D-12947 PF-IOO-PMC-01 Mars Pathfinder ProJect PLANETARY CONSTANTS AND MODELS Mars at Launch Earth at La_ Dec 2, 1996 SunI, Earth at Mars at Aniva July 4, 1997 December 1995 JilL Jet Propulsion Laboratory California Institute of Technology Pasadena, California JPL D-I2947 PF-IOO-PMC-OI Mars Pathfinder Project PLANETARY CONSTANTS AND MODELS Prepared by: ' Robin Vaughan December 1995 JPL Jet Propulsion Laboratory California Institute of Technology Pasadena, California JPL D-12947 PFolOO-PMCoO1 Revision History Date Changes Status 10/95 Issued preliminary version for review by project person-el Draft 12/95 First official release; incorporated comments from prelimi- Final nary review. JPL D- !294 7 PFo 100-PMCo 01 Contents List of Figures V 4 List of Tables vii List of Acronyms and Abbreviations ix INTRODUCTION 1 1.1 Purpose ............ .......................... 1 1.2 Scope .................... ................... 1 2 COORDINATE SYSTEMS 3 2.1 Definitions 2.1.1 Frame • " • • • ° * ° • " ° • * • ° ° " ° ° * ° • • • ° " " " ' • • • • • • 3 2.1.2 Center • " " • ° " " " ' ° ° ° • * " • • " • " • • " • " " " ° • ° • • • . • 6 2.1.3 Type .................................... 7 2.2 Celestial Systems . ; ............................. 7 2.2.1 The Inertial Reference Frames ..................... 8 2.2.2 Sun-Centered Systems .......................... 9 2.2.3 Earth-Centered Systems ......................... 9 2.2.4 Mars-Centered Systems ......................... 11 2.2.5 Spacecraft-CenteredSystems ...................... 16 2.2.6 MiscellaneousSystems -

The Artist As Researcher by Janneke Wesseling
Barcode at rear See it Sayit TheArt ist as Researcher Janneke Wesseling (ed.) Antennae Valiz, Amsterdam With contributions by Jeroen Boomgaard Jeremiah Day Siebren de Haan Stephan Dillemuth Irene Fortuyn Gijs Frieling Hadley+Maxwell Henri Jacobs WJMKok AglaiaKonrad Frank Mandersloot Aemout Mik Ruchama Noorda Va nessa Ohlraun Graeme Sullivan Moniek To ebosch Lonnie van Brummelen Hilde Van Gelder Philippe Va n Snick Barbara Visser Janneke Wesseling KittyZijlm ans Italo Zuffi See it Again, Say it Again: The Artist as Researcher Jan n eke We sse1 ing (ed.) See it Again, Say it Again: .. .. l TheArdst as Researcher Introduction Janneke We sseling The Use and Abuse of .. ...... .. .. .. .. 17 Research fo r Art and Vic e Versa Jeremiah Day .Art Research . 23 Hilde Va n Gelder Visual Contribution ................. 41 Ae rnout Mik Absinthe and Floating Tables . 51 Frank Mandersloot The Chimera ofMedtod ...... ........ ... 57 Jero en Boomgaard Reform and Education .. ...... ...... .. 73 Ruchama Noorda TheArtist as Researcher . 79 New Roles for New Realities Graeme Sullivan Visual Contribution .. .. .... .. .... .. 103 Ag laia Ko nrad Some Thoughts about Artistic . 117 Research Lonnie van Brummelen & Siebren de Haan Surtace Research . 123 Henri Jacobs TheArtist's To olbox .......... .......... 169 Irene Fortuyn Knight's Move . 175 The Idiosyncrasies of Artistic Research Ki tty Z ij 1 m a n s In Leaving the Shelter ................. 193 It alo Zu ffi Letterto Janneke Wesse6ng .......... 199 Va nessa Ohlraun Painting in Times of Research ..... 205 G ij s F r i e 1 ing Visual Contribution ........ .... ...... 211 Philippe Va n Snick TheAcademy and ........................... 221 the Corporate Pub&c Stephan Dillemuth Fleeting Profundity . 243 Mon iek To e bosch And, And , And So On . -

Optical Micromachines for Biological Studies
micromachines Review Optical Micromachines for Biological Studies Philippa-Kate Andrew 1 , Martin A. K. Williams 2,3 and Ebubekir Avci 1,3,* 1 Department of Mechanical and Electrical Engineering, Massey University, Palmerston North 4410, New Zealand; [email protected] 2 School of Fundamental Sciences, Massey University, Palmerston North 4410, New Zealand; [email protected] 3 MacDiarmid Institute for Advanced Materials and Nanotechnology, Wellington 6140, New Zealand * Correspondence: [email protected] Received: 21 January 2020; Accepted: 9 February 2020; Published: 13 February 2020 Abstract: Optical tweezers have been used for biological studies since shortly after their inception. However, over the years research has suggested that the intense laser light used to create optical traps may damage the specimens being studied. This review aims to provide a brief overview of optical tweezers and the possible mechanisms for damage, and more importantly examines the role of optical micromachines as tools for biological studies. This review covers the achievements to date in the field of optical micromachines: improvements in the ability to produce micromachines, including multi-body microrobots; and design considerations for both optical microrobots and the optical trapping set-up used for controlling them are all discussed. The review focuses especially on the role of micromachines in biological research, and explores some of the potential that the technology has in this area. Keywords: optical tweezers; multi-component micromanipulators; radiation damage; life sciences; optical microrobots 1. Introduction Improvements in tools for the visualisation of objects at the micro- and nano- scale have given researchers the ability to investigate materials and processes previously out of reach. -

Development and Characterization of an Atmospheric Pressure Ionization Source Matrix-Assisted Laser Desorption Electrospray Ioni
ABSTRACT SAMPSON, JASON SCOTT. Development and Characterization of an Atmospheric Pressure Ionization Source Matrix-Assisted Laser Desorption Electrospray Ionization Coupled to Fourier Transform Ion Cyclotron Resonance Mass Spectrometry for Analysis of Biological Macromolecules. (Under the direction of David Charles Muddiman). The field of mass spectrometry has grown tremendously over the past few decades due in large part to the continued application to new and interesting areas of exploration. The advent of soft ionization sources such as electrospray ionization and matrix-assisted laser desorption/ionization has dramatically increased the applications of mass spectrometry; in particular, analysis of complex biological samples. Electrospray ionization has demonstrated the capability to generate multiply-charged ions which increases the amount of information garnered from each analysis. Matrix-assisted laser desorption/ionization has demonstrated complex sample analysis requiring minimal sample preparation, which results in high throughput. As a result of these findings, there has been tremendous growth in the development of new ionization source technology in recent years for reducing sample preparation required prior to analysis for high throughput sample analysis and the generation of multiply-charged ions. Demonstrated herein is the development and characterization of an atmospheric pressure ionization source called matrix-assisted laser desorption electrospray ionization (MALDESI). MALDESI is a hybrid combination of MALDI and ESI which utilizes laser desorption with electrospray postionization for the generation of multiply-charged ions. Multiply-charged ions are of particular importance when using Fourier transform mass spectrometry, due to the increase in resolving power and mass accuracy with increasing charge on the molecule. Positive identification of biological macromolecules is demonstrated utilizing top-down characterization of intact polypeptides as well as high mass accuracy utilizing internal calibration. -

Local Lunar Gravity Field Analysis Over the South Poleaitken Basin from SELENE Farside Tracking Data
JOURNAL OF GEOPHYSICAL RESEARCH, VOL. 117, E02005, doi:10.1029/2011JE003831, 2012 Local lunar gravity field analysis over the South Pole-Aitken basin from SELENE farside tracking data Sander Goossens,1,2,3 Yoshiaki Ishihara,1 Koji Matsumoto,1 and Sho Sasaki1 Received 16 March 2011; revised 22 November 2011; accepted 13 December 2011; published 14 February 2012. [1] We present a method with which we determined the local lunar gravity field model over the South Pole-Aitken (SPA) basin on the farside of the Moon by estimating adjustments to a global lunar gravity field model using SELENE tracking data. Our adjustments are expressed in localized functions concentrated over the SPA region in a spherical cap with a radius of 45° centered at (191.1°E, 53.2°S), and the resolution is equivalent to a 150th degree and order spherical harmonics expansion. The new solution over SPA was used in several applications of geophysical analysis. It shows an increased correlation with high-resolution lunar topography in the frequency band l =40–70, and admittance values are slightly different and more leveled when compared to other, global gravity field models using the same data. The adjustments expressed in free-air anomalies and differences in Bouguer anomalies between the local solution and the a priori global solution correlate with topographic surface features. The Moho structure beneath the SPA basin is slightly modified in our solution, most notably at the southern rim of the Apollo basin and around the Zeeman crater. Citation: Goossens, S., Y. Ishihara, K. Matsumoto, and S. Sasaki (2012), Local lunar gravity field analysis over the South Pole-Aitken basin from SELENE farside tracking data, J. -

With Femtosecond Lasers, 572-74, 573/ Hydrodynamic Motion
Index Ablation Central hot spark ignition, by direct drive of ceramics, 216-21 implosion, 380-83 dielectric function during, 192-93, 193/ Central ignition scheme, 377/ 378 with femtosecond lasers, 572-74, 573/ Ceramics, ultrashort laser pulse interactions hydrodynamic motion role in, 2, 5-6 with, 216-21,217/218/ initial temperature at threshold of, 3 Coherent anti-Stokes Raman scattering measurement of dynamics of, 190, 191/ (CARS), 198, 206-8, 207/ of polymers, 225-26, 225/ 226/ 281-95 Coherent control of polymers with EUV laser, 535^0, 536/ raman spectroscopy, 206-8, 207/ 537/ 538/ 538<, 539/ of ultrashort laser pulse, 203^, 203/ spectral control of, 204-5, 205/ 206/ Collisional ionization, of dielectrics, 26-27 stages of, 68 69, 69/ Cosine model, of laser ionization, 110-14, Ablative cleaning, 37 110/115/ Ablative laser propulsion, 408-11, 409/ Coulomb explosion (CE) Artworks. See Painted artworks criterion for, 20-21, 21/ Asymptotic density profile, 12, 13/ features of, 18 from ultrashort laser pulse, 17-32 Band structure, 99 experimental evidence for, 18-20 Biological scaffold structures, from Crater ORMOCER®s, 149-51, 150/ 151/ formation of, 7-8, If Boltzmann plot, of plume, 77, 77/ hydrodynamic motion formation of, 4 Brillouin zone (BZ), of laser ionization, Crystal expansion, ultrashort laser pulse for, 100-108, 101/ 108/ 110 1-15 Bubble formation conclusion of, 14-15 calculation of, 254-57 hydrodynamic motion characteristics, 4—6 cavitation bubble dynamics, 262-67, 263/ hydrodynamic simulation, 11-14 264/ 265/ interference during, 9-11,10/ pulse trains with energies above threshold introduction to, 1-3 for, 271-72 shell and crater formation, 7-8, If pulse trains with energies below threshold for, 269-71,269/ Desorption ionization on silicon (DIOS) stress-induced, 254—67 MALDIv., 518-19, 518/521/ threshold for, 261-62 asSLDI, 506-7, 515-19 BZ.