Biophysical Studies of the G-Protein Coupled Neurotensin Receptor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

From Inverse Agonism to 'Paradoxical Pharmacology' Richard A
International Congress Series 1249 (2003) 27-37 From inverse agonism to 'Paradoxical Pharmacology' Richard A. Bond*, Kenda L.J. Evans, Zsirzsanna Callaerts-Vegh Department of Pharmacological and Pharmaceutical Sciences, University of Houston, 521 Science and Research Bldg 2, 4800 Caltioun, Houston, TX 77204-5037, USA Received 16 April 2003; accepted 16 April 2003 Abstract The constitutive or spontaneous activity of G protein-coupled receptors (GPCRs) and compounds acting as inverse agonists is a recent but well-established phenomenon. Dozens of receptor subtypes for numerous neurotransmitters and hormones have been shown to posses this property. However, do to the apparently low percentage of receptors in the spontaneously active state, the physiologic relevance of these findings remains questionable. The possibility that the reciprocal nature of the effects of agonists and inverse agonists may extend to cellular signaling is discussed, and that this may account for the beneficial effects of certain p-adrenoceptor inverse agonists in the treatment of heart failure. © 2003 Elsevier Science B.V. All rights reserved. Keywords. Inverse agonism; GPCR; Paradoxical pharmacology 1. Brief history of inverse agonism at G protein-coupled receptors For approximately three-quarters of a century, ligands that interacted with G protein- coupled receptors (GPCRs) were classified either as agonists or antagonists. Receptors were thought to exist in a single quiescent state that could only induce cellular signaling upon agonist binding to the receptor to produce an activated state of the receptor. In this model, antagonists had no cellular signaling ability on their own, but did bind to the receptor and prevented agonists from being able to bind and activate the receptor. -
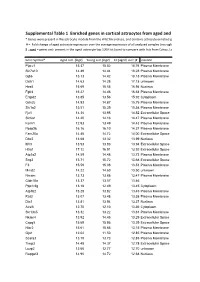
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

Guthrie Cdna Resource Center
cDNA Resource Center cDNA Resource Center Catalog cDNA Resource Center Missouri University of Science and Technology 400 W 11th Rolla, MO 65409 TEL: (573) 341-7610 FAX: (573) 341-7609 EMAIL: [email protected] www.cdna.org September, 2008 1 cDNA Resource Center Visit our web site for product updates 2 cDNA Resource Center The cDNA Resource Center The cDNA Resource Center is a service provided by the faculty of the Department of Biological Sciences of Missouri University of Science and Technology. The purpose of the cDNA Resource Center is to further scientific investigation by providing cDNA clones of human proteins involved in signal transduction processes. This is achieved by providing high quality clones for important signaling proteins in a timely manner. By high quality, we mean that the clones are • Sequence verified • Propagated in a versatile vector useful in bacterial and mammalian systems • Free of extraneous 3' and 5' untranslated regions • Expression verified (in most cases) by coupled in vitro transcription/translation assays • Available in wild-type, epitope-tagged and common mutant forms (e.g., constitutively- active or dominant negative) By timely, we mean that the clones are • Usually shipped within a day from when you place your order. Clones can be ordered from our web pages, by FAX or by phone. Within the United States, clones are shipped by overnight courier (FedEx); international orders are shipped International Priority (FedEx). The clones are supplied for research purposes only. Details on use of the material are included on the Material Transfer Agreement (page 3). Clones are distributed by agreement in Invitrogen's pcDNA3.1+ vector. -

Differential Gene Expression in Oligodendrocyte Progenitor Cells, Oligodendrocytes and Type II Astrocytes
Tohoku J. Exp. Med., 2011,Differential 223, 161-176 Gene Expression in OPCs, Oligodendrocytes and Type II Astrocytes 161 Differential Gene Expression in Oligodendrocyte Progenitor Cells, Oligodendrocytes and Type II Astrocytes Jian-Guo Hu,1,2,* Yan-Xia Wang,3,* Jian-Sheng Zhou,2 Chang-Jie Chen,4 Feng-Chao Wang,1 Xing-Wu Li1 and He-Zuo Lü1,2 1Department of Clinical Laboratory Science, The First Affiliated Hospital of Bengbu Medical College, Bengbu, P.R. China 2Anhui Key Laboratory of Tissue Transplantation, Bengbu Medical College, Bengbu, P.R. China 3Department of Neurobiology, Shanghai Jiaotong University School of Medicine, Shanghai, P.R. China 4Department of Laboratory Medicine, Bengbu Medical College, Bengbu, P.R. China Oligodendrocyte precursor cells (OPCs) are bipotential progenitor cells that can differentiate into myelin-forming oligodendrocytes or functionally undetermined type II astrocytes. Transplantation of OPCs is an attractive therapy for demyelinating diseases. However, due to their bipotential differentiation potential, the majority of OPCs differentiate into astrocytes at transplanted sites. It is therefore important to understand the molecular mechanisms that regulate the transition from OPCs to oligodendrocytes or astrocytes. In this study, we isolated OPCs from the spinal cords of rat embryos (16 days old) and induced them to differentiate into oligodendrocytes or type II astrocytes in the absence or presence of 10% fetal bovine serum, respectively. RNAs were extracted from each cell population and hybridized to GeneChip with 28,700 rat genes. Using the criterion of fold change > 4 in the expression level, we identified 83 genes that were up-regulated and 89 genes that were down-regulated in oligodendrocytes, and 92 genes that were up-regulated and 86 that were down-regulated in type II astrocytes compared with OPCs. -

The Anti-Apoptotic Role of Neurotensin
Cells 2013, 2, 124-135; doi:10.3390/cells2010124 OPEN ACCESS cells ISSN 2073-4409 www.mdpi.com/journal/cells Review The Anti-Apoptotic Role of Neurotensin Christelle Devader, Sophie Béraud-Dufour, Thierry Coppola and Jean Mazella * Institut de Pharmacologie Moléculaire et Cellulaire, CNRS UMR 7275, Université de Nice-Sophia Antipolis, 660 route des Lucioles, Valbonne 06560, France; E-Mails: [email protected] (C.D.); [email protected] (S.B.-D.); [email protected] (T.C.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +33-4-93-95-77-61; Fax: +33-4-93-95-77-08. Received: 24 January 2013; in revised form: 15 February 2013 / Accepted: 26 February 2013 / Published: 4 March 2013 Abstract: The neuropeptide, neurotensin, exerts numerous biological functions, including an efficient anti-apoptotic role, both in the central nervous system and in the periphery. This review summarizes studies that clearly evidenced the protective effect of neurotensin through its three known receptors. The pivotal involvement of the neurotensin receptor-3, also called sortilin, in the molecular mechanisms of the anti-apoptotic action of neurotensin has been analyzed in neuronal cell death, in cancer cell growth and in pancreatic beta cell protection. The relationships between the anti-apoptotic role of neurotensin and important physiological and pathological contexts are discussed in this review. Keywords: neurotensin; receptor; apoptosis; sortilin 1. Introduction The tridecapeptide neurotensin (NT) was isolated from bovine hypothalami on the basis of its ability to induce vasodilatation [1]. NT is synthesized from a precursor protein following excision by prohormone convertases [2]. -

The Role of Neurotensin and Its Receptors in Non-Gastrointestinal Cancers
Nikolaou et al. Cell Communication and Signaling (2020) 18:68 https://doi.org/10.1186/s12964-020-00569-y REVIEW Open Access The role of Neurotensin and its receptors in non-gastrointestinal cancers: a review Stella Nikolaou1,2,3, Shengyang Qiu1,3, Francesca Fiorentino3, Constantinos Simillis1, Shahnawaz Rasheed1,2,3, Paris Tekkis1,2,3 and Christos Kontovounisios1,2,3* Abstract Background: Neurotensin, originally isolated in 1973 has both endocrine and neuromodulator activity and acts through its three main receptors. Their role in promoting tumour cell proliferation, migration, DNA synthesis has been studied in a wide range of cancers. Expression of Neurotensin and its receptors has also been correlated to prognosis and prediction to treatment. Main body: The effects of NT are mediated through mitogen-activated protein kinases, epidermal growth factor receptors and phosphatidylinositol-3 kinases amongst others. This review is a comprehensive summary of the molecular pathways by which Neurotensin and its receptors act in cancer cells. Conclusion: Identifying the role of Neurotensin in the underlying molecular mechanisms in various cancers can give way to developing new agnostic drugs and personalizing treatment according to the genomic structure of various cancers. Keywords: Neurotensin, Neurotensin receptors, Cancer Background Neurotensin receptor 1(NTSR1), NTSR2 and NTSR3 (or Neurotensin (NT) was first detected by Carraway and Lee- sortilin 1) [6]. The first two are seven-transmembrane G- man in bovine hypothalamus in 1973 [1]. It is a 13-amino protein coupled receptors and the latter is a single trans- acid regulatory peptide present in both the central nervous membrane domain sorting receptor [6]. system (hypothalamus and pituitary) and the gastrointes- The NT gene transcription is transiently expressed in tinal tract (N cells of the jejunum and ileum) [1, 2]. -

Sortilin in Glucose Homeostasis: from Accessory Protein to Key Player? Nicolas Blondeau, Sophie Béraud-Dufour, Patricia Lebrun, Céline Hivelin, Thierry Coppola
Sortilin in Glucose Homeostasis: From Accessory Protein to Key Player? Nicolas Blondeau, Sophie Béraud-Dufour, Patricia Lebrun, Céline Hivelin, Thierry Coppola To cite this version: Nicolas Blondeau, Sophie Béraud-Dufour, Patricia Lebrun, Céline Hivelin, Thierry Coppola. Sortilin in Glucose Homeostasis: From Accessory Protein to Key Player?. Frontiers in Pharmacology, Frontiers, 2019, 9, 10.3389/fphar.2018.01561. hal-02265199 HAL Id: hal-02265199 https://hal.archives-ouvertes.fr/hal-02265199 Submitted on 5 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License fphar-09-01561 January 11, 2019 Time: 17:5 # 1 MINI REVIEW published: 15 January 2019 doi: 10.3389/fphar.2018.01561 Sortilin in Glucose Homeostasis: From Accessory Protein to Key Player? Nicolas Blondeau†, Sophie Béraud-Dufour†, Patricia Lebrun, Céline Hivelin and Thierry Coppola* Centre National de la Recherche Scientifique, Institut de Pharmacologie Moléculaire et Cellulaire, UMR 7275, Université Côte d’Azur, Valbonne, France The pharmacological properties and physiological roles of the type I receptor sortilin, also called neurotensin receptor-3, are various and complex. Sortilin is involved in important biological functions from neurotensin and pro-Nerve Growth Factor signaling in the central nervous system to regulation of glucose and lipid homeostasis in the periphery. -

Supplementary Table 2: Infection-Sensitive Genes
Supplementary Table 2: Infection-Sensitive Genes Supplementary Table 2: Upregulated with Adenoviral Infection (pages 1 - 7)- genes that were significant by ANOVA as well as significantly increased in control compared to both 'infected control' (Ad-LacZ) and 'calcineurin infected' (Ad-aCaN). Downregulated with Adenoviral Infection (pages 7 - 13) as above except for direction of change. Columns: Probe Set- Affymetrix probe set identifier for RG-U34A microarray, Symbol and Title- annotated information for above probe set (annotation downloaded June, 2004), ANOVA- p-value for 1-way Analysis of Variance, Uninfected, Ad-LacZ and Ad- aCaN- mean ± SEM expression for uninfected control, adenovirus mediated LacZ treated control, and adenovirus mediated calcineurin treated cultures respectively. Upregulated with Adenoviral Infection Probe Set Symbol Title ANOVA Uninfected Ad-LacZ Ad-aCaN Z48444cds_at Adam10 a disintegrin and metalloprotease domain 10 0.00001 837 ± 31 1107 ± 24 1028 ± 29 L26986_at Adcy8 adenylyl cyclase 8 0.00028 146 ± 12 252 ± 21 272 ± 21 U94340_at Adprt ADP-ribosyltransferase 1 0.00695 2096 ± 100 2783 ± 177 2586 ± 118 U01914_at Akap8 A kinase anchor protein 8 0.00080 993 ± 44 1194 ± 65 1316 ± 44 AB008538_at Alcam activated leukocyte cell adhesion molecule 0.01012 2775 ± 136 3580 ± 216 3429 ± 155 M34176_s_at Ap2b1 adaptor-related protein complex 2, beta 1 subunit 0.01389 2408 ± 199 2947 ± 143 3071 ± 116 D44495_s_at Apex1 apurinic/apyrimidinic endonuclease 1 0.00089 4959 ± 185 5816 ± 202 6057 ± 158 U16245_at Aqp5 aquaporin 5 0.02710 -

Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells Amy Sue Bogard University of Tennessee Health Science Center
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 12-2013 Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells Amy Sue Bogard University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Medical Cell Biology Commons, and the Medical Molecular Biology Commons Recommended Citation Bogard, Amy Sue , "Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells" (2013). Theses and Dissertations (ETD). Paper 330. http://dx.doi.org/10.21007/etd.cghs.2013.0029. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells Document Type Dissertation Degree Name Doctor of Philosophy (PhD) Program Biomedical Sciences Track Molecular Therapeutics and Cell Signaling Research Advisor Rennolds Ostrom, Ph.D. Committee Elizabeth Fitzpatrick, Ph.D. Edwards Park, Ph.D. Steven Tavalin, Ph.D. Christopher Waters, Ph.D. DOI 10.21007/etd.cghs.2013.0029 Comments Six month embargo expired June 2014 This dissertation is available at UTHSC Digital Commons: https://dc.uthsc.edu/dissertations/330 Adenylyl Cyclase 2 Selectively Regulates IL-6 Expression in Human Bronchial Smooth Muscle Cells A Dissertation Presented for The Graduate Studies Council The University of Tennessee Health Science Center In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy From The University of Tennessee By Amy Sue Bogard December 2013 Copyright © 2013 by Amy Sue Bogard. -

Single-Cell Transcriptomic Evidence for Dense Intracortical Neuropeptide
RESEARCH ARTICLE Single-cell transcriptomic evidence for dense intracortical neuropeptide networks Stephen J Smith1*, Uygar Su¨ mbu¨ l1, Lucas T Graybuck1, Forrest Collman1, Sharmishtaa Seshamani1, Rohan Gala1, Olga Gliko1, Leila Elabbady1, Jeremy A Miller1, Trygve E Bakken1, Jean Rossier2, Zizhen Yao1, Ed Lein1, Hongkui Zeng1, Bosiljka Tasic1, Michael Hawrylycz1* 1Allen Institute for Brain Science, Seattle, United States; 2Neuroscience Paris Seine, Sorbonne Universite´, Paris, France Abstract Seeking new insights into the homeostasis, modulation and plasticity of cortical synaptic networks, we have analyzed results from a single-cell RNA-seq study of 22,439 mouse neocortical neurons. Our analysis exposes transcriptomic evidence for dozens of molecularly distinct neuropeptidergic modulatory networks that directly interconnect all cortical neurons. This evidence begins with a discovery that transcripts of one or more neuropeptide precursor (NPP) and one or more neuropeptide-selective G-protein-coupled receptor (NP-GPCR) genes are highly abundant in all, or very nearly all, cortical neurons. Individual neurons express diverse subsets of NP signaling genes from palettes encoding 18 NPPs and 29 NP-GPCRs. These 47 genes comprise 37 cognate NPP/NP-GPCR pairs, implying the likelihood of local neuropeptide signaling. Here, we use neuron-type-specific patterns of NP gene expression to offer specific, testable predictions regarding 37 peptidergic neuromodulatory networks that may play prominent roles in cortical homeostasis and plasticity. *For correspondence: [email protected] (SJS); [email protected] (MH) Introduction Neuromodulation - the graded and relatively slow adjustment of fast synapse and ion channel func- Competing interests: The tion via diffusible cell-cell signaling molecules - is a fundamental requirement for adaptive nervous authors declare that no system function (Abbott and Regehr, 2004; Bargmann, 2012; Bucher and Marder, 2013; competing interests exist. -

Neurotensin Receptor 2 Is Induced in Astrocytes and Brain Endothelial
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.29.166637; this version posted June 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Neurotensin receptor 2 is induced in astrocytes and brain endothelial cells in relation to status epilepticus and neuroinflammation following pilocarpine administration in rats Running title : Induced astroglial NTSR2 in epilepsy Kyriatzis Grigorios1, Bernard Anne1, Bôle Angélique1, Pflieger Guillaume1, Chalas Petros1, Masse Maxime2, Lécorché Pascaline2, Jacquot Guillaume2, Ferhat Lotfi1* and Khrestchatisky Michel1* 1) Aix-Marseille Univ, CNRS, INP, Inst Neurophysiopathol, Marseille, France 2) VECT-HORUS, Faculté de Médecine, 27 Bd Jean Moulin, 13385 Marseille Cedex 5, France * Equally last and corresponding authors: [email protected] and [email protected] WORD COUNT Abstract: 242 Introduction: 919 Materials and Methods: 3511 Results: 4965 Discussion: 3146 References: 4183 Figure legends: 2262 MAIN POINTS Epilepsy is associated with increased NTSR2 expression in astrocytes and endothelial cells. Proinflammatory factors induce NTSR2 in astroglia with immediate early gene response. A neurotensin analog down regulates NTSR2 and glial cell inflammation. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.29.166637; this version posted June 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. ACKNOWLEDGEMENTS This work was supported by a fellowship to GK from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement No 642881, and AMIDEX (ICN PhD Program, ANR-11-IDEX-0001-02 grant) funded by the French Government «Investissements d’Avenir» program. -

Mediators and Receptors of Chronic Itch in Primates and Humans
MEDIATORS AND RECEPTORS OF CHRONIC ITCH IN PRIMATES AND HUMANS A Dissertation Submitted to the Temple University Graduate Board In Partial Fulfillment of the Requirements for the Degree DOCTOR OF PHILOSOPHY by Leigh Ann Nattkemper December 2015 Examining Committee Members: Gil Yosipovitch, MD, Advisory Chair, Department of Dermatology Mary Barbe, PhD, Department of Anatomy and Cell Biology Liselotte Jensen, PhD, Department of Microbiology and Immunology Alan Cowan, PhD, Department of Pharmacology Mark Hoon, PhD, External Member, National Institutes of Health (NIDCR) © Copyright 2015 by Leigh Nattkemper All Rights Reserved ii ABSTRACT Chronic itch has a significant impact on quality of life for millions of patients worldwide, on a level comparable to that of chronic pain. Yet, although there are a host of effective drugs available for pain, there are no therapies that specifically target chronic itch. Current experimental approaches to investigate the pathogenesis of chronic pruritus and to test novel therapeutic agents are largely limited to rodent models. However, rodent models display significant dermatological, neurophysiological, and immunological differences from humans with chronic itch. The disadvantages of the current rodent paradigms call for the design of a valid primate model of chronic itch. For four years, we have monitored scratching behavior in a primate colony (n=35) of Cynomolgus macaques ( Macaca fascicularis ) suffering from idiopathic chronic itch. By comparing molecular and genetic analyses of the primates’ skin to their quantified scratching behavior, we attempted to characterize the underlying mechanisms of chronic itch in this model. Furthermore, the expression of itch-related proteins was examined in both the primate model and in humans with pruritic diseases.