From Inverse Agonism to 'Paradoxical Pharmacology' Richard A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TABLE 1 Studies of Antagonist Activity in Constitutively Active
TABLE 1 Studies of antagonist activity in constitutively active receptors systems shown to demonstrate inverse agonism for at least one ligand Targets are natural Gs and constitutively active mutants (CAM) of GPCRs. Of 380 antagonists, 85% of the ligands demonstrate inverse agonism. Receptor Neutral Antagonist Inverse Agonist Reference Human β2-adrenergic Dichloroisoproterenol, pindolol, labetolol, timolol, Chidiac et al., 1996; Azzi et alprenolol, propranolol, ICI 118,551, cyanopindolol al., 2001 Turkey erythrocyte β-adrenergic Propranolol, pindolol Gotze et al., 1994 Human β2-adrenergic (CAM) Propranolol Betaxolol, ICI 118,551, sotalol, timolol Samama et al., 1994; Stevens and Milligan, 1998 Human/guinea pig β1-adrenergic Atenolol, propranolol Mewes et al., 1993 Human β1-adrenergic Carvedilol CGP20712A, metoprolol, bisoprolol Engelhardt et al., 2001 Rat α2D-adrenergic Rauwolscine, yohimbine, WB 4101, idazoxan, Tian et al., 1994 phentolamine, Human α2A-adrenergic Napthazoline, Rauwolscine, idazoxan, altipamezole, levomedetomidine, Jansson et al., 1998; Pauwels MPV-2088 (–)RX811059, RX 831003 et al., 2002 Human α2C-adrenergic RX821002, yohimbine Cayla et al., 1999 Human α2D-adrenergic Prazosin McCune et al., 2000 Rat α2-adrenoceptor MK912 RX821002 Murrin et al., 2000 Porcine α2A adrenoceptor (CAM- Idazoxan Rauwolscine, yohimbine, RX821002, MK912, Wade et al., 2001 T373K) phentolamine Human α2A-adrenoceptor (CAM) Dexefaroxan, (+)RX811059, (–)RX811059, RS15385, yohimbine, Pauwels et al., 2000 atipamezole fluparoxan, WB 4101 Hamster α1B-adrenergic -
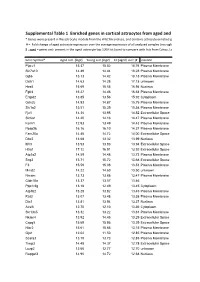
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

(12) Patent Application Publication (10) Pub. No.: US 2010/0221245 A1 Kunin (43) Pub
US 2010O221245A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2010/0221245 A1 Kunin (43) Pub. Date: Sep. 2, 2010 (54) TOPICAL SKIN CARE COMPOSITION Publication Classification (51) Int. Cl. (76) Inventor: Audrey Kunin, Mission Hills, KS A 6LX 39/395 (2006.01) (US) A6II 3L/235 (2006.01) A638/16 (2006.01) Correspondence Address: (52) U.S. Cl. ......................... 424/133.1: 514/533: 514/12 HUSCH BLACKWELL SANDERS LLP (57) ABSTRACT 4801 Main Street, Suite 1000 - KANSAS CITY, MO 64112 (US) The present invention is directed to a topical skin care com position. The composition has the unique ability to treat acne without drying out the user's skin. In particular, the compo (21) Appl. No.: 12/395,251 sition includes a base, an antibacterial agent, at least one anti-inflammatory agent, and at least one antioxidant. The (22) Filed: Feb. 27, 2009 antibacterial agent may be benzoyl peroxide. US 2010/0221 245 A1 Sep. 2, 2010 TOPCAL SKIN CARE COMPOSITION stay of acne treatment since the 1950s. Skin irritation is the most common side effect of benzoyl peroxide and other anti BACKGROUND OF THE INVENTION biotic usage. Some treatments can be severe and can leave the 0001. The present invention generally relates to composi user's skin excessively dry. Excessive use of some acne prod tions and methods for producing topical skin care. Acne Vul ucts may cause redness, dryness of the face, and can actually garis, or acne, is a common skin disease that is prevalent in lead to more acne. Therefore, it would be beneficial to provide teenagers and young adults. -

Ciproxifan, a Histamine H3 Receptor Antagonist, Reversibly Inhibits Monoamine Oxidase a and B Received: 05 September 2016 S
www.nature.com/scientificreports OPEN Ciproxifan, a histamine H3 receptor antagonist, reversibly inhibits monoamine oxidase A and B Received: 05 September 2016 S. Hagenow1, A. Stasiak2, R. R. Ramsay3 & H. Stark1 Accepted: 07 December 2016 Ciproxifan is a well-investigated histamine H receptor (H3R) inverse agonist/antagonist, showing Published: 13 January 2017 3 an exclusively high species-specific affinity at rodent compared to human H3R. It is well studied as reference compound for H3R in rodent models for neurological diseases connected with neurotransmitter dysregulation, e.g. attention deficit hyperactivity disorder or Alzheimer’s disease. In a screening for potential monoamine oxidase A and B inhibition ciproxifan showed efficacy on both enzyme isoforms. Further characterization of ciproxifan revealed IC50 values in a micromolar concentration range for human and rat monoamine oxidases with slight preference for monoamine oxidase B in both species. The inhibition by ciproxifan was reversible for both human isoforms. Regarding inhibitory potency of ciproxifan on rat brain MAO, these findings should be considered, when using high doses in rat models for neurological diseases. As the H3R and monoamine oxidases are all capable of affecting neurotransmitter modulation in brain, we consider dual targeting ligands as interesting approach for treatment of neurological disorders. Since ciproxifan shows only moderate activity at human targets, further investigations in animals are not of primary interest. On the other hand, it may serve as starting point for the development of dual targeting ligands. Ciproxifan (cyclopropyl 4-(3-(1H-imidazol-4-yl)propyloxy)phenyl methanone) is a well characterized species-specific histamine 3H receptor (H3R) inverse agonist/antagonist (Fig. -

Histamine Receptors
Tocris Scientific Review Series Tocri-lu-2945 Histamine Receptors Iwan de Esch and Rob Leurs Introduction Leiden/Amsterdam Center for Drug Research (LACDR), Division Histamine is one of the aminergic neurotransmitters and plays of Medicinal Chemistry, Faculty of Sciences, Vrije Universiteit an important role in the regulation of several (patho)physiological Amsterdam, De Boelelaan 1083, 1081 HV, Amsterdam, The processes. In the mammalian brain histamine is synthesised in Netherlands restricted populations of neurons that are located in the tuberomammillary nucleus of the posterior hypothalamus.1 Dr. Iwan de Esch is an assistant professor and Prof. Rob Leurs is These neurons project diffusely to most cerebral areas and have full professor and head of the Division of Medicinal Chemistry of been implicated in several brain functions (e.g. sleep/ the Leiden/Amsterdam Center of Drug Research (LACDR), VU wakefulness, hormonal secretion, cardiovascular control, University Amsterdam, The Netherlands. Since the seventies, thermoregulation, food intake, and memory formation).2 In histamine receptor research has been one of the traditional peripheral tissues, histamine is stored in mast cells, eosinophils, themes of the division. Molecular understanding of ligand- basophils, enterochromaffin cells and probably also in some receptor interaction is obtained by combining pharmacology specific neurons. Mast cell histamine plays an important role in (signal transduction, proliferation), molecular biology, receptor the pathogenesis of various allergic conditions. After mast cell modelling and the synthesis and identification of new ligands. degranulation, release of histamine leads to various well-known symptoms of allergic conditions in the skin and the airway system. In 1937, Bovet and Staub discovered compounds that antagonise the effect of histamine on these allergic reactions.3 Ever since, there has been intense research devoted towards finding novel ligands with (anti-) histaminergic activity. -

Making Sense of Pharmacology: Inverse Agonism and Functional Selectivity Kelly A
International Journal of Neuropsychopharmacology (2018) 21(10): 962–977 doi:10.1093/ijnp/pyy071 Advance Access Publication: August 6, 2018 Review review Making Sense of Pharmacology: Inverse Agonism and Functional Selectivity Kelly A. Berg and William P. Clarke Department of Pharmacology, University of Texas Health, San Antonio, Texas. Correspondence: William P. Clarke, PhD, Department of Pharmacology, Mail Stop 7764, UT Health at San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229 ([email protected]). Abstract Constitutive receptor activity/inverse agonism and functional selectivity/biased agonism are 2 concepts in contemporary pharmacology that have major implications for the use of drugs in medicine and research as well as for the processes of new drug development. Traditional receptor theory postulated that receptors in a population are quiescent unless activated by a ligand. Within this framework ligands could act as agonists with various degrees of intrinsic efficacy, or as antagonists with zero intrinsic efficacy. We now know that receptors can be active without an activating ligand and thus display “constitutive” activity. As a result, a new class of ligand was discovered that can reduce the constitutive activity of a receptor. These ligands produce the opposite effect of an agonist and are called inverse agonists. The second topic discussed is functional selectivity, also commonly referred to as biased agonism. Traditional receptor theory also posited that intrinsic efficacy is a single drug property independent of the system in which the drug acts. However, we now know that a drug, acting at a single receptor subtype, can have multiple intrinsic efficacies that differ depending on which of the multiple responses coupled to a receptor is measured. -

In Vitro Pharmacology of Clinically Used Central Nervous System-Active Drugs As Inverse H1 Receptor Agonists
0022-3565/07/3221-172–179$20.00 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS Vol. 322, No. 1 Copyright © 2007 by The American Society for Pharmacology and Experimental Therapeutics 118869/3215703 JPET 322:172–179, 2007 Printed in U.S.A. In Vitro Pharmacology of Clinically Used Central Nervous System-Active Drugs as Inverse H1 Receptor Agonists R. A. Bakker,1 M. W. Nicholas,2 T. T. Smith, E. S. Burstein, U. Hacksell, H. Timmerman, R. Leurs, M. R. Brann, and D. M. Weiner Department of Medicinal Chemistry, Leiden/Amsterdam Center for Drug Research, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands (R.A.B., H.T., R.L.); ACADIA Pharmaceuticals Inc., San Diego, California (R.A.B., M.W.N., T.T.S., E.S.B., U.H., M.R.B., D.M.W.); and Departments of Pharmacology (M.R.B.), Neurosciences (D.M.W.), and Psychiatry (D.M.W.), University of California, San Diego, California Received January 2, 2007; accepted March 30, 2007 Downloaded from ABSTRACT The human histamine H1 receptor (H1R) is a prototypical G on this screen, we have reported on the identification of 8R- protein-coupled receptor and an important, well characterized lisuride as a potent stereospecific partial H1R agonist (Mol target for the development of antagonists to treat allergic con- Pharmacol 65:538–549, 2004). In contrast, herein we report on jpet.aspetjournals.org ditions. Many neuropsychiatric drugs are also known to po- a large number of varied clinical and chemical classes of drugs tently antagonize this receptor, underlying aspects of their side that are active in the central nervous system that display potent effect profiles. -

Guthrie Cdna Resource Center
cDNA Resource Center cDNA Resource Center Catalog cDNA Resource Center Missouri University of Science and Technology 400 W 11th Rolla, MO 65409 TEL: (573) 341-7610 FAX: (573) 341-7609 EMAIL: [email protected] www.cdna.org September, 2008 1 cDNA Resource Center Visit our web site for product updates 2 cDNA Resource Center The cDNA Resource Center The cDNA Resource Center is a service provided by the faculty of the Department of Biological Sciences of Missouri University of Science and Technology. The purpose of the cDNA Resource Center is to further scientific investigation by providing cDNA clones of human proteins involved in signal transduction processes. This is achieved by providing high quality clones for important signaling proteins in a timely manner. By high quality, we mean that the clones are • Sequence verified • Propagated in a versatile vector useful in bacterial and mammalian systems • Free of extraneous 3' and 5' untranslated regions • Expression verified (in most cases) by coupled in vitro transcription/translation assays • Available in wild-type, epitope-tagged and common mutant forms (e.g., constitutively- active or dominant negative) By timely, we mean that the clones are • Usually shipped within a day from when you place your order. Clones can be ordered from our web pages, by FAX or by phone. Within the United States, clones are shipped by overnight courier (FedEx); international orders are shipped International Priority (FedEx). The clones are supplied for research purposes only. Details on use of the material are included on the Material Transfer Agreement (page 3). Clones are distributed by agreement in Invitrogen's pcDNA3.1+ vector. -

Differential Gene Expression in Oligodendrocyte Progenitor Cells, Oligodendrocytes and Type II Astrocytes
Tohoku J. Exp. Med., 2011,Differential 223, 161-176 Gene Expression in OPCs, Oligodendrocytes and Type II Astrocytes 161 Differential Gene Expression in Oligodendrocyte Progenitor Cells, Oligodendrocytes and Type II Astrocytes Jian-Guo Hu,1,2,* Yan-Xia Wang,3,* Jian-Sheng Zhou,2 Chang-Jie Chen,4 Feng-Chao Wang,1 Xing-Wu Li1 and He-Zuo Lü1,2 1Department of Clinical Laboratory Science, The First Affiliated Hospital of Bengbu Medical College, Bengbu, P.R. China 2Anhui Key Laboratory of Tissue Transplantation, Bengbu Medical College, Bengbu, P.R. China 3Department of Neurobiology, Shanghai Jiaotong University School of Medicine, Shanghai, P.R. China 4Department of Laboratory Medicine, Bengbu Medical College, Bengbu, P.R. China Oligodendrocyte precursor cells (OPCs) are bipotential progenitor cells that can differentiate into myelin-forming oligodendrocytes or functionally undetermined type II astrocytes. Transplantation of OPCs is an attractive therapy for demyelinating diseases. However, due to their bipotential differentiation potential, the majority of OPCs differentiate into astrocytes at transplanted sites. It is therefore important to understand the molecular mechanisms that regulate the transition from OPCs to oligodendrocytes or astrocytes. In this study, we isolated OPCs from the spinal cords of rat embryos (16 days old) and induced them to differentiate into oligodendrocytes or type II astrocytes in the absence or presence of 10% fetal bovine serum, respectively. RNAs were extracted from each cell population and hybridized to GeneChip with 28,700 rat genes. Using the criterion of fold change > 4 in the expression level, we identified 83 genes that were up-regulated and 89 genes that were down-regulated in oligodendrocytes, and 92 genes that were up-regulated and 86 that were down-regulated in type II astrocytes compared with OPCs. -

Title Page CEP-26401 (Irdabisant), a Potent and Selective Histamine H3 Receptor Antagonist/ Inverse Agonist with Cognition-Enhan
JPET Fast Forward. Published on October 14, 2011 as DOI: 10.1124/jpet.111.186585 JPET FastThis articleForward. has not Published been copyedited on and October formatted. 14,The final2011 version as DOI:10.1124/jpet.111.186585 may differ from this version. Title Page CEP-26401 (Irdabisant), a potent and selective histamine H3 receptor antagonist/ inverse agonist with cognition-enhancing and wake promoting activities Downloaded from Rita Raddatz, Robert L. Hudkins, Joanne R. Mathiasen*, John A. Gruner*, Dorothy G. Flood*, Lisa D. Aimone, Siyuan Le*, Hervé Schaffhauser*, Maciej Gasior*, Donna jpet.aspetjournals.org Bozyczko-Coyne, Michael J. Marino*, Mark A. Ator, Edward R. Bacon, John P. Mallamo and Michael Williams* at ASPET Journals on September 24, 2021 Discovery Research Cephalon, Inc. 145 Brandywine Parkway West Chester, PA 19380 USA Copyright 2011 by the American Society for Pharmacology and Experimental Therapeutics. JPET Fast Forward. Published on October 14, 2011 as DOI: 10.1124/jpet.111.186585 This article has not been copyedited and formatted. The final version may differ from this version. JPET #186585 2 Running Title H3 Receptor Antagonist CEP-26401 Corresponding author: Rita Raddatz Cephalon, Inc. 145 Brandywine Parkway West Chester, PA 19380 USA PHONE : (610) 738-6728 FAX: (610) 738-6305 Downloaded from [email protected] Text Pages: 48 Tables: 6 jpet.aspetjournals.org Figures: 7 References: 40 at ASPET Journals on September 24, 2021 Abstract (# of words): 240 Introduction (# of words): 490 Discussion (# of words): 1500 Abbreviations: H3R, histamine H3 receptor; CEP-26401, 6-{4-[3-((R)-2-methyl- pyrrolidin-1-yl)-propoxy]-phenyl}-2H-pyridazin-3-one HCl; ABT-239, 4-(2-{2-[(2R)-2- methyl-1-pyrrolidinyl]ethyl}-1-benzofuran-5-yl)benzonitrile (L) tartrate; RAMH, R-α- methylhistamine; GTPγS, guanosine 5`-(γ-thio)triphosphate; PPI, prepulse inhibition; [3H]NAMH, [3H]N-α-methylhistamine; RID, ratio of investigation duration; DTT, dithiothreitol; EEG, electroencephalographic; EMG, electromyographic; SWS, slow wave sleep; REMS, rapid eye movement sleep. -

Product Information
Print Date: Oct 31st 2017 Product Information www.tocris.com Product Name: Histamine H3 Receptor Tocriset™ Catalog No.: 1876 Batch No.: 1 1. Tocriset™ Description A Tocriset™ consists of 3 to 5 key compounds that are active within a defined pharmacological area or a signaling pathway. Most compounds are supplied in a solid format in a specified molar amount so that solvent can be added directly to the vial. For example, addition of 500 μL of solvent to a vial containing 5 μmol of compound yields a 10 mM stock solution. Some compounds that are unsuitable for lyophilization are provided pre-dissolved in DMSO. The Histamine H3 Receptor Tocriset™ contains the listed products as lyophilised solids which can be used to study the pharmacology of the histamine H3 receptor. Cat.No. Product / Activity Batch Amount Format 0569 (R)-(-)-α-Methylhistamine dihydrobromide 7 5 μmol Freeze-dried solid Potent, selective H3 agonist 0644 Thioperamide 7 5 μmol Freeze-dried solid H3 antagonist, active in vivo 0729 Imetit dihydrobromide 2 5 μmol Freeze-dried solid Standard selective H3 agonist 0752 Clobenpropit dihydrobromide 2 5 μmol Freeze-dried solid Highly potent, selective H3 antagonist 0779 Iodophenpropit dihydrobromide 2 5 μmol Freeze-dried solid Potent, selective H3 antagonist 2. Storage & Solubility SOLIDS: Provided storage is as stated on the product label and the vial is kept tightly sealed, the product can be stored for up to 6 months from date of receipt. SOLUTIONS: We recommend that stock solutions, once prepared, are stored aliquoted in tightly sealed vials at -20°C or below and used within 1 month, unless indicated below. -

Cannabinoid-1 Receptor Inverse Agonists: Current Understanding of Mechanism of Action and Unanswered Questions
International Journal of Obesity (2009) 33, 947–955 & 2009 Macmillan Publishers Limited All rights reserved 0307-0565/09 $32.00 www.nature.com/ijo REVIEW Cannabinoid-1 receptor inverse agonists: current understanding of mechanism of action and unanswered questions TM Fong1 and SB Heymsfield2 1Merck Research Laboratories, Department of Metabolic Disorders, Rahway, NJ, USA and 2Merck Research Laboratories, Global Center of Scientific Affairs, Rahway, NJ, USA Rimonabant and taranabant are two extensively studied cannabinoid-1 receptor (CB1R) inverse agonists. Their effects on in vivo peripheral tissue metabolism are generally well replicated. The central nervous system site of action of taranabant or rimonabant is firmly established based on brain receptor occupancy studies. At the whole-body level, the mechanism of action of CB1R inverse agonists includes a reduction in food intake and an increase in energy expenditure. At the tissue level, fat mass reduction, liver lipid reduction and improved insulin sensitivity have been shown. These effects on tissue metabolism are readily explained by CB1R inverse agonist acting on brain CB1R and indirectly influencing the tissue metabolism through the autonomic nervous system. It has also been hypothesized that rimonabant acts directly on adipocytes, hepatocytes, pancreatic islets or skeletal muscle in addition to acting on brain CB1R, although strong support for the contribution of peripherally located CB1R to in vivo efficacy is still lacking. This review will carefully examine the published literature