Single-Cell Transcriptomic Evidence for Dense Intracortical Neuropeptide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

From Inverse Agonism to 'Paradoxical Pharmacology' Richard A
International Congress Series 1249 (2003) 27-37 From inverse agonism to 'Paradoxical Pharmacology' Richard A. Bond*, Kenda L.J. Evans, Zsirzsanna Callaerts-Vegh Department of Pharmacological and Pharmaceutical Sciences, University of Houston, 521 Science and Research Bldg 2, 4800 Caltioun, Houston, TX 77204-5037, USA Received 16 April 2003; accepted 16 April 2003 Abstract The constitutive or spontaneous activity of G protein-coupled receptors (GPCRs) and compounds acting as inverse agonists is a recent but well-established phenomenon. Dozens of receptor subtypes for numerous neurotransmitters and hormones have been shown to posses this property. However, do to the apparently low percentage of receptors in the spontaneously active state, the physiologic relevance of these findings remains questionable. The possibility that the reciprocal nature of the effects of agonists and inverse agonists may extend to cellular signaling is discussed, and that this may account for the beneficial effects of certain p-adrenoceptor inverse agonists in the treatment of heart failure. © 2003 Elsevier Science B.V. All rights reserved. Keywords. Inverse agonism; GPCR; Paradoxical pharmacology 1. Brief history of inverse agonism at G protein-coupled receptors For approximately three-quarters of a century, ligands that interacted with G protein- coupled receptors (GPCRs) were classified either as agonists or antagonists. Receptors were thought to exist in a single quiescent state that could only induce cellular signaling upon agonist binding to the receptor to produce an activated state of the receptor. In this model, antagonists had no cellular signaling ability on their own, but did bind to the receptor and prevented agonists from being able to bind and activate the receptor. -
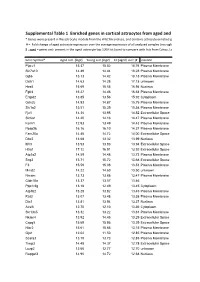
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

Neurotensin Activates Gabaergic Interneurons in the Prefrontal Cortex
The Journal of Neuroscience, February 16, 2005 • 25(7):1629–1636 • 1629 Behavioral/Systems/Cognitive Neurotensin Activates GABAergic Interneurons in the Prefrontal Cortex Kimberly A. Petrie,1 Dennis Schmidt,1 Michael Bubser,1 Jim Fadel,1 Robert E. Carraway,2 and Ariel Y. Deutch1 1Departments of Pharmacology and Psychiatry, Vanderbilt University Medical Center, Nashville, Tennessee 37212, and 2Department of Physiology, University of Massachusetts Medical Center, Worcester, Massachusetts 01655 Converging data suggest a dysfunction of prefrontal cortical GABAergic interneurons in schizophrenia. Morphological and physiological studies indicate that cortical GABA cells are modulated by a variety of afferents. The peptide transmitter neurotensin may be one such modulator of interneurons. In the rat prefrontal cortex (PFC), neurotensin is exclusively localized to dopamine axons and has been suggested to be decreased in schizophrenia. However, the effects of neurotensin on cortical interneurons are poorly understood. We used in vivo microdialysis in freely moving rats to assess whether neurotensin regulates PFC GABAergic interneurons. Intra-PFC administra- tion of neurotensin concentration-dependently increased extracellular GABA levels; this effect was impulse dependent, being blocked by treatment with tetrodotoxin. The ability of neurotensin to increase GABA levels in the PFC was also blocked by pretreatment with 2-[1-(7-chloro-4-quinolinyl)-5-(2,6-dimethoxyphenyl)pyrazole-3-yl)carbonylamino]tricyclo(3.3.1.1.3.7)decan-2-carboxylic acid (SR48692), a high-affinity neurotensin receptor 1 (NTR1) antagonist. This finding is consistent with our observation that NTR1 was localized to GABAergic interneurons in the PFC, particularly parvalbumin-containing interneurons. Because neurotensin is exclusively localized to dopamine axons in the PFC, we also determined whether neurotensin plays a role in the ability of dopamine agonists to increase extracellular GABA levels. -

Edinburgh Research Explorer
Edinburgh Research Explorer International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list Citation for published version: Davenport, AP, Alexander, SPH, Sharman, JL, Pawson, AJ, Benson, HE, Monaghan, AE, Liew, WC, Mpamhanga, CP, Bonner, TI, Neubig, RR, Pin, JP, Spedding, M & Harmar, AJ 2013, 'International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands', Pharmacological reviews, vol. 65, no. 3, pp. 967-86. https://doi.org/10.1124/pr.112.007179 Digital Object Identifier (DOI): 10.1124/pr.112.007179 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Pharmacological reviews Publisher Rights Statement: U.S. Government work not protected by U.S. copyright General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 1521-0081/65/3/967–986$25.00 http://dx.doi.org/10.1124/pr.112.007179 PHARMACOLOGICAL REVIEWS Pharmacol Rev 65:967–986, July 2013 U.S. -

Molecular Mechanisms Involved in the Regulation of Agouti-Related Peptide and Neuropeptide Y by Endocrine Disrupting Chemical Bisphenol a in Hypothalamic Neurons
Molecular mechanisms involved in the regulation of agouti-related peptide and neuropeptide Y by endocrine disrupting chemical bisphenol A in hypothalamic neurons by Neruja Loganathan A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy Department of Physiology University of Toronto © Copyright by Neruja Loganathan 2021 Molecular mechanisms involved in the regulation of agouti-related peptide and neuropeptide Y by endocrine disrupting chemical bisphenol A in hypothalamic neurons Neruja Loganathan Doctor of Philosophy Department of Physiology University of Toronto 2021 Abstract Bisphenol A (BPA), a ubiquitous endocrine disrupting chemical found in plastics and receipts, is a disruptor of reproductive function and is a known ‘obesogen’ as it is linked to increased body mass index in humans and leads to weight gain in animal models. The hypothalamus houses orexigenic NPY/AgRP neurons, which integrate peripheral hormones and nutritional signals, to increase food intake and decrease energy expenditure. NPY neurons are also afferent regulators of the hypothalamic-pituitary gonadal axis, and thus reproductive function. This thesis investigated whether the NPY/AgRP neurons, and particularly Npy and Agrp expression, are altered by BPA. We hypothesized that BPA increases Npy and Agrp gene expression in hypothalamic neurons and that this effect is mediated through nuclear receptor activation, induction of cellular stress and subsequent transcription factor activation or circadian dysregulation. We demonstrated that BPA increased Agrp mRNA expression in mHypoA-59 and mHypoE-41 cells. Inhibition of AMPK and knock-down of transcription factor ATF3 prevented the BPA-mediated increase in Agrp expression in the mHypoA-59 cells. ATF3 was also required for BPA-mediated increase in Npy in the mHypoE-41 cells. -

Guthrie Cdna Resource Center
cDNA Resource Center cDNA Resource Center Catalog cDNA Resource Center Missouri University of Science and Technology 400 W 11th Rolla, MO 65409 TEL: (573) 341-7610 FAX: (573) 341-7609 EMAIL: [email protected] www.cdna.org September, 2008 1 cDNA Resource Center Visit our web site for product updates 2 cDNA Resource Center The cDNA Resource Center The cDNA Resource Center is a service provided by the faculty of the Department of Biological Sciences of Missouri University of Science and Technology. The purpose of the cDNA Resource Center is to further scientific investigation by providing cDNA clones of human proteins involved in signal transduction processes. This is achieved by providing high quality clones for important signaling proteins in a timely manner. By high quality, we mean that the clones are • Sequence verified • Propagated in a versatile vector useful in bacterial and mammalian systems • Free of extraneous 3' and 5' untranslated regions • Expression verified (in most cases) by coupled in vitro transcription/translation assays • Available in wild-type, epitope-tagged and common mutant forms (e.g., constitutively- active or dominant negative) By timely, we mean that the clones are • Usually shipped within a day from when you place your order. Clones can be ordered from our web pages, by FAX or by phone. Within the United States, clones are shipped by overnight courier (FedEx); international orders are shipped International Priority (FedEx). The clones are supplied for research purposes only. Details on use of the material are included on the Material Transfer Agreement (page 3). Clones are distributed by agreement in Invitrogen's pcDNA3.1+ vector. -

Differential Gene Expression in Oligodendrocyte Progenitor Cells, Oligodendrocytes and Type II Astrocytes
Tohoku J. Exp. Med., 2011,Differential 223, 161-176 Gene Expression in OPCs, Oligodendrocytes and Type II Astrocytes 161 Differential Gene Expression in Oligodendrocyte Progenitor Cells, Oligodendrocytes and Type II Astrocytes Jian-Guo Hu,1,2,* Yan-Xia Wang,3,* Jian-Sheng Zhou,2 Chang-Jie Chen,4 Feng-Chao Wang,1 Xing-Wu Li1 and He-Zuo Lü1,2 1Department of Clinical Laboratory Science, The First Affiliated Hospital of Bengbu Medical College, Bengbu, P.R. China 2Anhui Key Laboratory of Tissue Transplantation, Bengbu Medical College, Bengbu, P.R. China 3Department of Neurobiology, Shanghai Jiaotong University School of Medicine, Shanghai, P.R. China 4Department of Laboratory Medicine, Bengbu Medical College, Bengbu, P.R. China Oligodendrocyte precursor cells (OPCs) are bipotential progenitor cells that can differentiate into myelin-forming oligodendrocytes or functionally undetermined type II astrocytes. Transplantation of OPCs is an attractive therapy for demyelinating diseases. However, due to their bipotential differentiation potential, the majority of OPCs differentiate into astrocytes at transplanted sites. It is therefore important to understand the molecular mechanisms that regulate the transition from OPCs to oligodendrocytes or astrocytes. In this study, we isolated OPCs from the spinal cords of rat embryos (16 days old) and induced them to differentiate into oligodendrocytes or type II astrocytes in the absence or presence of 10% fetal bovine serum, respectively. RNAs were extracted from each cell population and hybridized to GeneChip with 28,700 rat genes. Using the criterion of fold change > 4 in the expression level, we identified 83 genes that were up-regulated and 89 genes that were down-regulated in oligodendrocytes, and 92 genes that were up-regulated and 86 that were down-regulated in type II astrocytes compared with OPCs. -

Structure and Dynamics of a Constitutively Active Neurotensin Receptor Received: 26 August 2016 Brian E
www.nature.com/scientificreports OPEN Structure and dynamics of a constitutively active neurotensin receptor Received: 26 August 2016 Brian E. Krumm1,†, Sangbae Lee2, Supriyo Bhattacharya2, Istvan Botos3, Courtney F. White1, Accepted: 03 November 2016 Haijuan Du1, Nagarajan Vaidehi2 & Reinhard Grisshammer1 Published: 07 December 2016 Many G protein-coupled receptors show constitutive activity, resulting in the production of a second messenger in the absence of an agonist; and naturally occurring constitutively active mutations in receptors have been implicated in diseases. To gain insight into mechanistic aspects of constitutive activity, we report here the 3.3 Å crystal structure of a constitutively active, agonist-bound neurotensin receptor (NTSR1) and molecular dynamics simulations of agonist-occupied and ligand-free receptor. Comparison with the structure of a NTSR1 variant that has little constitutive activity reveals uncoupling of the ligand-binding domain from conserved connector residues, that effect conformational changes during GPCR activation. Furthermore, molecular dynamics simulations show strong contacts between connector residue side chains and increased flexibility at the intracellular receptor face as features that coincide with robust signalling in cells. The loss of correlation between the binding pocket and conserved connector residues, combined with altered receptor dynamics, possibly explains the reduced neurotensin efficacy in the constitutively active NTSR1 and a facilitated initial engagement with G protein in the absence of agonist. G protein-coupled receptors (GPCRs) are highly dynamic and versatile signalling molecules that mediate second messenger responses within the cell. Binding of an extracellular agonist causes conformational changes in the receptor, triggering activation of signalling partners such as G proteins or arrestin molecules on the intracellu- lar side of the membrane. -

Neuropeptide Y in the Amygdala Induces Long-Term
The Journal of Neuroscience, January 23, 2008 • 28(4):893–903 • 893 Behavioral/Systems/Cognitive Neuropeptide Y in the Amygdala Induces Long-Term Resilience to Stress-Induced Reductions in Social Responses But Not Hypothalamic–Adrenal–Pituitary Axis Activity or Hyperthermia Tammy J. Sajdyk,1 Philip L. Johnson,1 Randy J. Leitermann,3 Stephanie D. Fitz,1 Amy Dietrich,1 Michelle Morin,2 Donald R. Gehlert,2 Janice H. Urban,3 and Anantha Shekhar1 1Institute of Psychiatric Research, Department of Psychiatry, Indiana University School of Medicine, Indianapolis, Indiana 46202, 2Eli Lilly, Indianapolis, Indiana 46201, and 3Department of Physiology and Biophysics and Interdepartmental Neurosciences Program, Rosalind Franklin University of Medicine and Science, North Chicago, Illinois 60064 Resilience to mental and physical stress is a key determinant for the survival and functioning of mammals. Although the importance of stress resilience has been recognized, the underlying neural mediators have not yet been identified. Neuropeptide Y (NPY) is a peptide known for its anti-anxiety-like effects mediated via the amygdala. The results of our current study demonstrate, for the first time that repeated administration of NPY directly into the basolateral nucleus of the amygdala (BLA) produces selective stress-resilient behavioral responses to an acute restraint challenge as measured in the social interaction test, but has no effect on hypothalamic–adrenal–pituitary axisactivityorstress-inducedhyperthermia.Moreimportantly,theresilientbehaviorsobservedintheNPY-treatedanimalswerepresent for up to 8 weeks. Antagonizing the activity of calcineurin, a protein phosphatase involved in neuronal remodeling and present in NPY receptor containing neurons within the BLA, blocked the development of long-term, but not the acute increases in social interaction responses induced by NPY administration. -

Targeting Neuropeptide Receptors for Cancer Imaging and Therapy: Perspectives with Bombesin, Neurotensin, and Neuropeptide-Y Receptors
Journal of Nuclear Medicine, published on September 4, 2014 as doi:10.2967/jnumed.114.142000 CONTINUING EDUCATION Targeting Neuropeptide Receptors for Cancer Imaging and Therapy: Perspectives with Bombesin, Neurotensin, and Neuropeptide-Y Receptors Clément Morgat1–3, Anil Kumar Mishra2–4, Raunak Varshney4, Michèle Allard1,2,5, Philippe Fernandez1–3, and Elif Hindié1–3 1CHU de Bordeaux, Service de Médecine Nucléaire, Bordeaux, France; 2University of Bordeaux, INCIA, UMR 5287, Talence, France; 3CNRS, INCIA, UMR 5287, Talence, France; 4Division of Cyclotron and Radiopharmaceutical Sciences, Institute of Nuclear Medicine and Allied Sciences, DRDO, New Delhi, India; and 5EPHE, Bordeaux, France Learning Objectives: On successful completion of this activity, participants should be able to list and discuss (1) the presence of bombesin receptors, neurotensin receptors, or neuropeptide-Y receptors in some major tumors; (2) the perspectives offered by radiolabeled peptides targeting these receptors for imaging and therapy; and (3) the choice between agonists and antagonists for tumor targeting and the relevance of various PET radionuclides for molecular imaging. Financial Disclosure: The authors of this article have indicated no relevant relationships that could be perceived as a real or apparent conflict of interest. CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through October 2017. -

Neurotensin Receptor 1 Antagonist SR48692 Improves Response To
Published OnlineFirst August 8, 2017; DOI: 10.1158/1078-0432.CCR-17-0861 Cancer Therapy: Preclinical Clinical Cancer Research Neurotensin Receptor 1 Antagonist SR48692 Improves Response to Carboplatin by Enhancing Apoptosis and Inhibiting Drug Efflux in Ovarian Cancer Jin Liu1, Mikael€ Agopiantz2,3,Joel€ Poupon4, Zherui Wu1, Pierre-Alexandre Just5, Bruno Borghese6, Evelyne Segal-Bendirdjian 1, Guillaume Gauchotte3,7, Anne Gompel1,8, and Patricia Forgez1 Abstract Purpose: The high affinity receptor 1 (NTSR1) and its agonist, Results: SR48692 enhanced the response to carboplatin in neurotensin (NTS), are correlated with tumor cell aggressiveness ovarian cancer cells and experimental tumors. When SR48692 in most solid tumors. As chemoresistance and tumor aggres- is combined with carboplatin, we noted a major improvement siveness are often related, we decided to study the role of the of platinum-induced DNA damage and cell death, as well as a NTSR1 complex within platinum-based chemotherapy responses. decrease in tumor growth. The relationship of these results In an ovarian model, we studied carboplatin because it is the main to clinical studies was made by the detection of NTS and standard of care for ovarian cancer. NTSR1 in 72% and 74% of ovarian cancer, respectively. Fur- Experimental Design: Experimental tumors and in vitro studies thermore, in a large series of high-grade ovarian cancer, NTSR1 were performed using SKOV3 and A2780 cells treated with mRNA was shown to correlate with higher stages and platinum carboplatin, with or without a very specific NTSR1 antagonist, resistance. SR48692. We measured the effects of these treatments on cell Conclusions: This study strongly suggests that the addition apoptosis and apoptosis-related proteins, platinum accumula- of NTSR1 inhibitor in combination with platinum salt–based tion in the cell and nucleus, and the expression and localization of therapy will improve the response to the drug. -

Identification of Neuropeptide Receptors Expressed By
RESEARCH ARTICLE Identification of Neuropeptide Receptors Expressed by Melanin-Concentrating Hormone Neurons Gregory S. Parks,1,2 Lien Wang,1 Zhiwei Wang,1 and Olivier Civelli1,2,3* 1Department of Pharmacology, University of California Irvine, Irvine, California 92697 2Department of Developmental and Cell Biology, University of California Irvine, Irvine, California 92697 3Department of Pharmaceutical Sciences, University of California Irvine, Irvine, California 92697 ABSTRACT the MCH system or demonstrated high expression lev- Melanin-concentrating hormone (MCH) is a 19-amino- els in the LH and ZI, were tested to determine whether acid cyclic neuropeptide that acts in rodents via the they are expressed by MCH neurons. Overall, 11 neuro- MCH receptor 1 (MCHR1) to regulate a wide variety of peptide receptors were found to exhibit significant physiological functions. MCH is produced by a distinct colocalization with MCH neurons: nociceptin/orphanin population of neurons located in the lateral hypothala- FQ opioid receptor (NOP), MCHR1, both orexin recep- mus (LH) and zona incerta (ZI), but MCHR1 mRNA is tors (ORX), somatostatin receptors 1 and 2 (SSTR1, widely expressed throughout the brain. The physiologi- SSTR2), kisspeptin recepotor (KissR1), neurotensin cal responses and behaviors regulated by the MCH sys- receptor 1 (NTSR1), neuropeptide S receptor (NPSR), tem have been investigated, but less is known about cholecystokinin receptor A (CCKAR), and the j-opioid how MCH neurons are regulated. The effects of most receptor (KOR). Among these receptors, six have never classical neurotransmitters on MCH neurons have been before been linked to the MCH system. Surprisingly, studied, but those of most neuropeptides are poorly several receptors thought to regulate MCH neurons dis- understood.