Airway Inflammation in Mice Adam8 Limits the Development of Allergic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supporting Online Material
1 2 3 4 5 6 7 Supplementary Information for 8 9 Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic 10 retinopathy 11 12 *Samuel A. Mills, *Andrew I. Jobling, *Michael A. Dixon, Bang V. Bui, Kirstan A. Vessey, Joanna A. Phipps, 13 Ursula Greferath, Gene Venables, Vickie H.Y. Wong, Connie H.Y. Wong, Zheng He, Flora Hui, James C. 14 Young, Josh Tonc, Elena Ivanova, Botir T. Sagdullaev, Erica L. Fletcher 15 * Joint first authors 16 17 Corresponding author: 18 Prof. Erica L. Fletcher. Department of Anatomy & Neuroscience. The University of Melbourne, Grattan St, 19 Parkville 3010, Victoria, Australia. 20 Email: [email protected] ; Tel: +61-3-8344-3218; Fax: +61-3-9347-5219 21 22 This PDF file includes: 23 24 Supplementary text 25 Figures S1 to S10 26 Tables S1 to S7 27 Legends for Movies S1 to S2 28 SI References 29 30 Other supplementary materials for this manuscript include the following: 31 32 Movies S1 to S2 33 34 35 36 1 1 Supplementary Information Text 2 Materials and Methods 3 Microglial process movement on retinal vessels 4 Dark agouti rats were anaesthetized, injected intraperitoneally with rhodamine B (Sigma-Aldrich) to label blood 5 vessels and retinal explants established as described in the main text. Retinal microglia were labelled with Iba-1 6 and imaging performed on an inverted confocal microscope (Leica SP5). Baseline images were taken for 10 7 minutes, followed by the addition of PBS (10 minutes) and then either fractalkine or fractalkine + candesartan 8 (10 minutes) using concentrations outlined in the main text. -
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
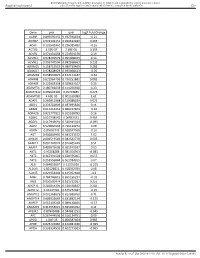
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

R&D Assay for Alzheimer's Disease
R&DR&D assayassay forfor Alzheimer’sAlzheimer’s diseasedisease Target screening⳼ Ⲽ㬔 antibody array, ᢜ⭉㬔 ⸽ἐⴐ Amyloid β-peptide Alzheimer’s disease⯸ ኸᷠ᧔ ᆹ⸽ inhibitor, antibody, ELISA kit Surwhrph#Surilohu#Dqwlerg|#Duud| 6OUSFBUFE 1."5SFBUFE )41 $3&# &3, &3, )41 $3&# &3, &3, 壤伡庰䋸TBNQMF ɅH 侴䋸嵄䍴䋸BOBMZUFT䋸䬱娴哜塵 1$ 1$ 1$ 1$ 5IFNPTUSFGFSFODFEBSSBZT 1$ 1$ QQ α 34, .4, 503 Q α 34, .4, 503 %SVHTDSFFOJOH0òUBSHFUFòFDUT0ATHWAY涭廐 6OUSFBUFE 堄币䋸4BNQMF侴䋸8FTUFSOPS&-*4"䍘䧽 1."5SFBUFE P 8FTUFSOCMPU廽喜儤应侴䋸0, Z 4VCTUSBUF -JHIU )31DPOKVHBUFE1BO "OUJQIPTQIPUZSPTJOF .FBO1JYFM%FOTJUZ Y $BQUVSF"OUJCPEZ 5BSHFU"OBMZUF "SSBZ.FNCSBOF $3&# &3, &3, )41 .4, Q α 34, 503 Human XL Cytokine Array kit (ARY022, 102 analytes) Adiponectin,Aggrecan,Angiogenin,Angiopoietin-1,Angiopoietin-2,BAFF,BDNF,Complement,Component C5/C5a,CD14,CD30,CD40L, Chitinase 3-like 1,Complement Factor D,C-Reactive Protein,Cripto-1,Cystatin C,Dkk-1,DPPIV,EGF,EMMPRIN,ENA-78,Endoglin, Fas L,FGF basic,FGF- 7,FGF-19,Flt-3 L,G-CSF,GDF-15,GM-CSF,GRO-α,Grow th Hormone,HGF,ICAM-1,IFN-γ,IGFBP-2,IGFBP-3, IL-1α,IL-1β, IL-1ra,IL-2,IL-3,IL-4,IL- 5,IL-6,IL-8, IL-10,IL-11,IL-12, IL-13,IL-15,IL-16,IL-17A,IL-18 BPa,IL-19,IL-22, IL-23,IL-24,IL-27, IL-31,IL-32α/β/γ,IL-33,IL-34,IP-10,I-TAC,Kallikrein 3,Leptin,LIF,Lipocalin-2,MCP-1,MCP-3,M-CSF,MIF,MIG,MIP-1α/MIP-1β,MIP-3α,MIP-3β,MMP-9, Myeloperoxidase,Osteopontin, p70, PDGF-AA, PDGF-AB/BB,Pentraxin-3, PF4, RAGE, RANTES,RBP4,Relaxin-2, Resistin,SDF-1α,Serpin E1, SHBG, ST2, TARC,TFF3,TfR,TGF- ,Thrombospondin-1,TNF-α, uPAR, VEGF, Vitamin D BP Human Protease (34 analytes) / -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -

ADAM8 Expression in Invasive Breast Cancer Promotes Tumor Dissemination and Metastasis
ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis Mathilde Romagnoli, Nora Mineva, Michael Polmear, Catharina Conrad, Srimathi Srinivasan, Delphine Loussouarn, Sophie Barillé-Nion, Irene Georgakoudi, Aine Dagg, Enda Mcdermott, et al. To cite this version: Mathilde Romagnoli, Nora Mineva, Michael Polmear, Catharina Conrad, Srimathi Srinivasan, et al.. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Molecular Medicine, Wiley Open Access, 2014, 6 (2), pp.278-294. 10.1002/emmm.201303373. inserm-02447040 HAL Id: inserm-02447040 https://www.hal.inserm.fr/inserm-02447040 Submitted on 21 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Research Article ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis Mathilde Romagnoli1, Nora D Mineva1, Michael Polmear2, Catharina Conrad3, Srimathi Srinivasan1, Delphine Loussouarn4, Sophie Barille-Nion4, Irene Georgakoudi2, Aine Dagg5,6, Enda W McDermott6, Michael J Duffy6, Patricia M. McGowan5,6, Uwe Schlomann3, Maddy Parsons7,Jorg€ W Bartsch3 & Gail E Sonenshein1,* Abstract Introduction The transmembrane metalloprotease-disintegrin ADAM8 mediates Cancer metastasis results from a multistep process that selects for cell adhesion and shedding of ligands, receptors and extracellular invasive tumor cells capable of escaping from the primary site and matrix components. -

ADAM10 Controls Collagen Signaling and Cell Migration on Collagen by Shedding the Ectodomain of Discoidin Domain Receptor 1 (DDR1)
M BoC | ARTICLE ADAM10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (DDR1) Yasuyuki Shitomia, Ida B. Thøgersenb, Noriko Itoa, Birgit Leitingerc, Jan J. Enghildb, and Yoshifumi Itoha aKennedy Institute of Rheumatology, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford OX3 7FY, United Kingdom; bDepartment of Molecular Biology and Genetics, University of Aarhus, DK-8000 Aarhus C, Denmark; cNational Heart and Lung Institute, Imperial College London, London SW7 2AZ, United Kingdom ABSTRACT Discoidin domain receptor 1 (DDR1) is a receptor tyrosine kinase that binds and Monitoring Editor transmits signals from various collagens in epithelial cells. However, how DDR1–dependent Jean E. Schwarzbauer signaling is regulated has not been understood. Here we report that collagen binding in- Princeton University duces ADAM10-dependent ectodomain shedding of DDR1. DDR1 shedding is not a result of Received: Oct 21, 2014 an activation of its signaling pathway, since DDR1 mutants defective in signaling were shed Revised: Dec 4, 2014 in an efficient manner. DDR1 and ADAM10 were found to be in a complex on the cell surface, Accepted: Dec 16, 2014 but shedding did not occur unless collagen bound to DDR1. Using a shedding-resistant DDR1 mutant, we found that ADAM10-dependent DDR1 shedding regulates the half-life of colla- gen-induced phosphorylation of the receptor. Our data also revealed that ADAM10 plays an important role in regulating DDR1-mediated cell adhesion to achieve efficient cell migration on collagen matrices. INTRODUCTION Extracellular matrix (ECM) is essential in multicellular organisms to discoidin domain receptors (DDRs), glycoprotein VI, leukocyte-asso- maintain functional tissue structures; it acts as scaffolding to support ciated, immunoglobulin-like receptors, and mannose receptors such cell migration and as a reservoir for growth factors. -

Strategies to Target ADAM17 in Disease: from Its Discovery to the Irhom Revolution
molecules Review Strategies to Target ADAM17 in Disease: From Its Discovery to the iRhom Revolution Matteo Calligaris 1,2,†, Doretta Cuffaro 2,†, Simone Bonelli 1, Donatella Pia Spanò 3, Armando Rossello 2, Elisa Nuti 2,* and Simone Dario Scilabra 1,* 1 Proteomics Group of Fondazione Ri.MED, Research Department IRCCS ISMETT (Istituto Mediterraneo per i Trapianti e Terapie ad Alta Specializzazione), Via E. Tricomi 5, 90145 Palermo, Italy; [email protected] (M.C.); [email protected] (S.B.) 2 Department of Pharmacy, University of Pisa, Via Bonanno 6, 56126 Pisa, Italy; [email protected] (D.C.); [email protected] (A.R.) 3 Università degli Studi di Palermo, STEBICEF (Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche), Viale delle Scienze Ed. 16, 90128 Palermo, Italy; [email protected] * Correspondence: [email protected] (E.N.); [email protected] (S.D.S.) † These authors contributed equally to this work. Abstract: For decades, disintegrin and metalloproteinase 17 (ADAM17) has been the object of deep investigation. Since its discovery as the tumor necrosis factor convertase, it has been considered a major drug target, especially in the context of inflammatory diseases and cancer. Nevertheless, the development of drugs targeting ADAM17 has been harder than expected. This has generally been due to its multifunctionality, with over 80 different transmembrane proteins other than tumor necrosis factor α (TNF) being released by ADAM17, and its structural similarity to other metalloproteinases. This review provides an overview of the different roles of ADAM17 in disease and the effects of its ablation in a number of in vivo models of pathological conditions. -

Novel Alternatively Spliced ADAM8 Isoforms Contribute to the Aggressive Bone Metastatic Phenotype of Lung Cancer
Oncogene (2010) 29, 3758–3769 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 www.nature.com/onc ORIGINAL ARTICLE Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer I Herna´ndez1, JL Moreno2, C Zandueta1, L Montuenga3,4 and F Lecanda1 1Adhesion and Metastasis Laboratory, Division of Oncology, University of Navarra, Pamplona, Spain; 2Department of Orthopaedics, University of Maryland School of Medicine, Baltimore, MD, USA; 3Department of Histology and Pathology, School of Medicine, University of Navarra, Pamplona, Spain and 4Biomarkers Laboratory, Center for Applied Biomedical Research (CIMA), University of Navarra, Pamplona, Spain ADAMs (a disintegrin and metalloprotease) are trans- survival rates for lung cancer are o15% in all developed membrane proteins involved in a variety of physiological countries. It is estimated that 30–40% of lung cancer processes and tumorigenesis. Recently, ADAM8 has been patients with advanced NSCLC suffer from bone associated with poor prognosis of lung cancer. However, metastasis (Coleman, 1997). Patients with bone metas- its contribution to tumorigenesis in the context of lung tasis experience pain, metabolic syndromes and spinal cancer metastasis remains unknown. Native ADAM8 cord compression associated with pathological fractures expression levels were lower in lung cancer cell lines. as a consequence of osteolytic lesions. In contrast, we identified and characterized two novel ADAMs (a disintegrin and metalloprotease) form a spliced isoforms encoding truncated proteins, D18a and large family of cell-surface proteins, which are char- D140, which were present in several tumor cell lines and not acterized by disintegrin and metalloproteinase domains, in normal cells. Overexpression of D18a protein resulted in that possess adhesive properties and proteolytic activ- enhanced invasive activity in vitro.ADAM8anditsD140 ities, respectively (Lu et al., 2007). -

NIK Controls Classical and Alternative NF-Kb Activation and Is Necessary for the Survival of Human T-Cell Lymphoma Cells
Published OnlineFirst March 27, 2013; DOI: 10.1158/1078-0432.CCR-12-3151 Clinical Cancer Human Cancer Biology Research NIK Controls Classical and Alternative NF-kB Activation and Is Necessary for the Survival of Human T-cell Lymphoma Cells Lina Odqvist1, Margarita Sanchez-Beato 1,3, Santiago Montes-Moreno1,10, Esperanza Martín-Sanchez 1, Raquel Pajares2, Lydia Sanchez-Verde 2, Pablo L. Ortiz-Romero4, Jose Rodriguez5, Socorro M. Rodríguez-Pinilla1,6, Francisca Iniesta-Martínez9, Juan Carlos Solera-Arroyo11, Rafael Ramos-Asensio12, Teresa Flores13, Javier Menarguez Palanca7, Federico García Bragado14, Purificacion Domínguez Franjo8, and Miguel A. Piris1,10 Abstract Purpose: Peripheral T-cell lymphomas (PTCL) are a heterogeneous entity of neoplasms with poor prognosis, a lack of effective therapies, and a largely unknown molecular pathology. Deregulated NF-kB activity has been associated with several lymphoproliferative diseases, but its importance in T-cell lymphomagenesis is poorly understood. We investigated the function of the NF-kB–inducing kinase (NIK), in this pathway and its role as a potential molecular target in T-cell lymphomas. Experimental Design: We used immunohistochemistry to analyze the expression of different NF-kB members in primary human PTCL samples and to study its clinical impact. With the aim of inhibiting the pathway, we used genetic silencing of NIK in several T-cell lymphoma cell lines and observed its effect on downstream targets and cell viability. Results: We showed that the NF-kB pathway was activated in a subset of PTCLs associated with poor overall survival. NIK was overexpressed in a number of PTCL cell lines and primary samples, and a pivotal role for NIK in the survival of these tumor cells was unveiled. -

Expression of Proteolytic Enzymes by Small Cell Lung Cancer Circulating Tumor Cell Lines
cancers Article Expression of Proteolytic Enzymes by Small Cell Lung Cancer Circulating Tumor Cell Lines Barbara Rath 1, Lukas Klameth 2, Adelina Plangger 1, Maximilian Hochmair 3, Ernst Ulsperger 4, Ihor Huk 1, Robert Zeillinger 5 and Gerhard Hamilton 1,* 1 Department of Vascular Surgery, Medical University of Vienna, A-1090 Vienna, Austria; [email protected] (B.R.); [email protected] (A.P.); [email protected] (I.H.) 2 Center for Pathophysiology, Infectiology and Immunology, Medical University of Vienna, A-1090 Vienna, Austria; [email protected] 3 Respiratory Oncology Unit, Otto Wagner Hospital, A-1140 Vienna, Austria; [email protected] 4 Hospital Horn, A-3580 Horn, Austria; [email protected] 5 Molecular Oncology Group, Department of Obstetrics and Gynecology, Comprehensive Cancer Center-Gynecological Cancer Unit, Medical University of Vienna, A-1090 Vienna, Austria; [email protected] * Correspondence: [email protected] Received: 28 December 2018; Accepted: 17 January 2019; Published: 19 January 2019 Abstract: Small cell lung cancer (SCLC) is an aggressive type of lung cancer which disseminates vigorously and has a dismal prognosis. Metastasis of SCLC is linked to an extremely high number of circulating tumor cells (CTCs), which form chemoresistant spheroids, termed tumorospheres. Intravasation and extravasation during tumor spread requires the activity of a number of proteases to disintegrate the stroma and vascular tissue. Generation of several permanent SCLC CTC lines allowed us to screen for the expression of 35 proteases using Western blot arrays. Cell culture supernatants of two CTC lines, namely BHGc7 and 10, were analyzed for secreted proteases, including matrix metalloproteinases (MMPs), ADAM/TS, cathepsins, kallikreins, and others, and compared to proteases expressed by SCLC cell lines (GLC14, GLC16, NCI-H526 and SCLC26A). -

A Potential Therapeutic Role for Angiotensin Converting Enzyme 2 in Human Pulmonary Arterial Hypertension
ERJ Express. Published on June 14, 2018 as doi: 10.1183/13993003.02638-2017 Early View Original article A potential therapeutic role for Angiotensin Converting Enzyme 2 in human pulmonary arterial hypertension Anna R. Hemnes, Anandharajan Rathinasabapathy, Eric A. Austin, Evan L. Brittain, Erica J. Carrier, Xinping Chen, Joshua P. Fessel, Candice D. Fike, Peter Fong, Niki Fortune, Robert E. Gerszten, Jennifer A. Johnson, Mark Kaplowitz, John H. Newman, Robert Piana, Meredith E. Pugh, Todd W. Rice, Ivan M. Robbins, Lisa Wheeler, Chang Yu, James E. Loyd, James West Please cite this article as: Hemnes AR, Rathinasabapathy A, Austin EA, et al. A potential therapeutic role for Angiotensin Converting Enzyme 2 in human pulmonary arterial hypertension. Eur Respir J 2018; in press (https://doi.org/10.1183/13993003.02638-2017). This manuscript has recently been accepted for publication in the European Respiratory Journal. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJ online. Copyright ©ERS 2018 Copyright 2018 by the European Respiratory Society. A potential therapeutic role for Angiotensin Converting Enzyme 2 in human pulmonary arterial hypertension Anna R. Hemnes, MD*1, Anandharajan Rathinasabapathy, PhD*1, Eric A. Austin, MD, MSCI2, Evan L. Brittain, MD, MSCI3, Erica J. Carrier, PhD1, Xinping Chen, PhD1, Joshua P. Fessel, MD, PhD1, Candice D. Fike, MD2, Peter Fong, MD3, Niki Fortune1, Robert E. Gerszten, MD4, Jennifer A. Johnson, MD1, Mark Kaplowitz2, John H. -

Endostatin and Irradiation Modifies the Activity of ADAM10 and Neprilysin in Breast Cancer Cells
MOLECULAR MEDICINE REPORTS 14: 2343-2351, 2016 Endostatin and irradiation modifies the activity of ADAM10 and neprilysin in breast cancer cells ESRA ARSLAN AYDEMIR1, ECE ŞIMŞEK2, AYLIN FIDAN KORCUM3 and KAYAHAN FIŞKIN2 1Department of Biology, Science Faculty; 2Department of Nutrition and Dietetics, Antalya School of Health; 3Department of Radiation Oncology, School of Medicine, Akdeniz University, Antalya 07058, Turkey Received December 10, 2015; Accepted June 6, 2016 DOI: 10.3892/mmr.2016.5463 Abstract. Angiogenesis, the formation of new blood vessels, and to discard of waste products. Inhibition angiogenesis is a is regarded as a key cancer cell property. Endostatin (ES) is promising strategy in cancer therapy (1,2). a potential antiangiogenic agent and it may be useful when Targeting angiogenesis may add to the therapeutic effect implemented in combination with other cancer therapeutic conventional cancer therapeutic strategies, including chemo- strategies. The present study investigated the in vitro effects of therapeutic agents and radiation therapy, as they are not yet ES, radiotherapy (RT) or combination therapy (ES + RT) on entirely effective against cancer. Angiogenesis is stringently two important proteases, a disintegrin and metalloproteinase controlled via the balance of pro-angiogenic and anti-angio- domain-containing protein 10 (ADAM10) and neprilysin genic factors, which are diffusible chemical signal molecules (NEP) in 4T1 mouse breast cancer cells and the more meta- secreted from tumor cells. Angiogenesis may be initiated by static phenotype of 4THMpc breast cancer cells. 4T1 and altering the net balance between positive and negative regulators 4THMpc cells were treated with recombinant murine ES via increased production of any one of the positive regulators (4 µg/ml) alone, RT (45 Gy) alone or with ES + RT.