ADAM10 Controls Collagen Signaling and Cell Migration on Collagen by Shedding the Ectodomain of Discoidin Domain Receptor 1 (DDR1)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Overexpresssion of Tyrosine Kinase Discoidin Domain Receptor I (DDR1 ) in Transitional Cell Carcinoma
Overexpresssion of Tyrosine Kinase Discoidin domain receptor I (DDR1 ) in Transitional cell Carcinoma Szu-Ting Chen,* and Shie-Liang Hsieh* Institute and Department of Microbiology and Immunology, National Yang-Ming University, Taipei, Taiwan; To whom proofs are to be sent: Shie-Liang Hsieh, Department and Institute of Microbiology and Immunology, National Yang-Ming University, Shih-Pai, Taipei 112, Taiwan. E-mail address: [email protected] Telephone number: 886-2-28267161 Fax number: 886-2-28277933 INTRODUCTION DDR1, discoidin domain receptor 1, belongs to the novel subfamily of tyrosine kinase receptor, which forms homodimer upon ligand engagement. DDR1 is distinguished from other receptor tyrosine kinase by the discoidin domain in their extracellular domain, which is a homology region originally identified in Dictyostelium discoideum (slime mold) protein, discoidin I, and involves in cells aggregation. Discoidin-1 has binding specificity toward galactose and N-acetyl galactosamine and is essential for slime mold cells adhesion, migration and aggregation during its development, suggesting that DDR1 shared similar biologically function in mammalian [1-3]. DDR1 has been found mainly distributed and in human tissue epithelia, such as kidney, breast, lung [3], bronchial [4] and keratinocytes [5]. Furthermore, DDR1 has been also reported its expression in immune system like the monocyte-derived dendritic cells, annotated as CD167 [6] and macrophage[7]. However, recently, the overexpression of DDR1 has been detected in several human cancers, such as primary breast cancer [1, 8, 9] ovarian[10, 11], brain[12], esophageal cancer [13], and TCC (our data unpublished) in which raising the possibility that DDR1 may play a important role in tumorigenesis [14]. -

Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse
Welcome to More Choice CD Marker Handbook For more information, please visit: Human bdbiosciences.com/eu/go/humancdmarkers Mouse bdbiosciences.com/eu/go/mousecdmarkers Human and Mouse CD Marker Handbook Human and Mouse CD Marker Key Markers - Human Key Markers - Mouse CD3 CD3 CD (cluster of differentiation) molecules are cell surface markers T Cell CD4 CD4 useful for the identification and characterization of leukocytes. The CD CD8 CD8 nomenclature was developed and is maintained through the HLDA (Human Leukocyte Differentiation Antigens) workshop started in 1982. CD45R/B220 CD19 CD19 The goal is to provide standardization of monoclonal antibodies to B Cell CD20 CD22 (B cell activation marker) human antigens across laboratories. To characterize or “workshop” the antibodies, multiple laboratories carry out blind analyses of antibodies. These results independently validate antibody specificity. CD11c CD11c Dendritic Cell CD123 CD123 While the CD nomenclature has been developed for use with human antigens, it is applied to corresponding mouse antigens as well as antigens from other species. However, the mouse and other species NK Cell CD56 CD335 (NKp46) antibodies are not tested by HLDA. Human CD markers were reviewed by the HLDA. New CD markers Stem Cell/ CD34 CD34 were established at the HLDA9 meeting held in Barcelona in 2010. For Precursor hematopoetic stem cell only hematopoetic stem cell only additional information and CD markers please visit www.hcdm.org. Macrophage/ CD14 CD11b/ Mac-1 Monocyte CD33 Ly-71 (F4/80) CD66b Granulocyte CD66b Gr-1/Ly6G Ly6C CD41 CD41 CD61 (Integrin b3) CD61 Platelet CD9 CD62 CD62P (activated platelets) CD235a CD235a Erythrocyte Ter-119 CD146 MECA-32 CD106 CD146 Endothelial Cell CD31 CD62E (activated endothelial cells) Epithelial Cell CD236 CD326 (EPCAM1) For Research Use Only. -

Supporting Online Material
1 2 3 4 5 6 7 Supplementary Information for 8 9 Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic 10 retinopathy 11 12 *Samuel A. Mills, *Andrew I. Jobling, *Michael A. Dixon, Bang V. Bui, Kirstan A. Vessey, Joanna A. Phipps, 13 Ursula Greferath, Gene Venables, Vickie H.Y. Wong, Connie H.Y. Wong, Zheng He, Flora Hui, James C. 14 Young, Josh Tonc, Elena Ivanova, Botir T. Sagdullaev, Erica L. Fletcher 15 * Joint first authors 16 17 Corresponding author: 18 Prof. Erica L. Fletcher. Department of Anatomy & Neuroscience. The University of Melbourne, Grattan St, 19 Parkville 3010, Victoria, Australia. 20 Email: [email protected] ; Tel: +61-3-8344-3218; Fax: +61-3-9347-5219 21 22 This PDF file includes: 23 24 Supplementary text 25 Figures S1 to S10 26 Tables S1 to S7 27 Legends for Movies S1 to S2 28 SI References 29 30 Other supplementary materials for this manuscript include the following: 31 32 Movies S1 to S2 33 34 35 36 1 1 Supplementary Information Text 2 Materials and Methods 3 Microglial process movement on retinal vessels 4 Dark agouti rats were anaesthetized, injected intraperitoneally with rhodamine B (Sigma-Aldrich) to label blood 5 vessels and retinal explants established as described in the main text. Retinal microglia were labelled with Iba-1 6 and imaging performed on an inverted confocal microscope (Leica SP5). Baseline images were taken for 10 7 minutes, followed by the addition of PBS (10 minutes) and then either fractalkine or fractalkine + candesartan 8 (10 minutes) using concentrations outlined in the main text. -

3 Cleavage Products of Notch 2/Site and Myelopoiesis by Dysregulating
ADAM10 Overexpression Shifts Lympho- and Myelopoiesis by Dysregulating Site 2/Site 3 Cleavage Products of Notch This information is current as David R. Gibb, Sheinei J. Saleem, Dae-Joong Kang, Mark of October 4, 2021. A. Subler and Daniel H. Conrad J Immunol 2011; 186:4244-4252; Prepublished online 2 March 2011; doi: 10.4049/jimmunol.1003318 http://www.jimmunol.org/content/186/7/4244 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2011/03/02/jimmunol.100331 Material 8.DC1 http://www.jimmunol.org/ References This article cites 45 articles, 16 of which you can access for free at: http://www.jimmunol.org/content/186/7/4244.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 4, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2011 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology ADAM10 Overexpression Shifts Lympho- and Myelopoiesis by Dysregulating Site 2/Site 3 Cleavage Products of Notch David R. -

ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease
International Journal of Molecular Sciences Review ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease Francesca Tosetti 1,* , Massimo Alessio 2, Alessandro Poggi 1,† and Maria Raffaella Zocchi 3,† 1 Molecular Oncology and Angiogenesis Unit, IRCCS Ospedale Policlinico S. Martino Largo R. Benzi 10, 16132 Genoa, Italy; [email protected] 2 Proteome Biochemistry, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] 3 Division of Immunology, Transplants and Infectious Diseases, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work as last author. Abstract: Enzymes, once considered static molecular machines acting in defined spatial patterns and sites of action, move to different intra- and extracellular locations, changing their function. This topological regulation revealed a close cross-talk between proteases and signaling events involving post-translational modifications, membrane tyrosine kinase receptors and G-protein coupled recep- tors, motor proteins shuttling cargos in intracellular vesicles, and small-molecule messengers. Here, we highlight recent advances in our knowledge of regulation and function of A Disintegrin And Metalloproteinase (ADAM) endopeptidases at specific subcellular sites, or in multimolecular com- plexes, with a special focus on ADAM10, and tumor necrosis factor-α convertase (TACE/ADAM17), since these two enzymes belong to the same family, share selected substrates and bioactivity. We will discuss some examples of ADAM10 activity modulated by changing partners and subcellular compartmentalization, with the underlying hypothesis that restraining protease activity by spatial Citation: Tosetti, F.; Alessio, M.; segregation is a complex and powerful regulatory tool. -

Cell-Autonomous FLT3L Shedding Via ADAM10 Mediates Conventional Dendritic Cell Development in Mouse Spleen
Cell-autonomous FLT3L shedding via ADAM10 mediates conventional dendritic cell development in mouse spleen Kohei Fujitaa,b,1, Svetoslav Chakarovc,1, Tetsuro Kobayashid, Keiko Sakamotod, Benjamin Voisind, Kaibo Duanc, Taneaki Nakagawaa, Keisuke Horiuchie, Masayuki Amagaib, Florent Ginhouxc, and Keisuke Nagaod,2 aDepartment of Dentistry and Oral Surgery, Keio University School of Medicine, Tokyo 160-8582, Japan; bDepartment of Dermatology, Keio University School of Medicine, Tokyo 160-8582, Japan; cSingapore Immunology Network, Agency for Science, Technology and Research, Biopolis, 138648 Singapore; dDermatology Branch, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892; and eDepartment of Orthopedic Surgery, National Defense Medical College, Tokorozawa 359-8513, Japan Edited by Kenneth M. Murphy, Washington University School of Medicine, St. Louis, MO, and approved June 10, 2019 (received for review November 4, 2018) Conventional dendritic cells (cDCs) derive from bone marrow (BM) intocDC1sorcDC2stakesplaceintheBM(3),andthese precursors that undergo cascades of developmental programs to pre-cDC1s and pre-cDC2s ultimately differentiate into cDC1s terminally differentiate in peripheral tissues. Pre-cDC1s and pre- and cDC2s after migrating to nonlymphoid and lymphoid tissues. + + cDC2s commit in the BM to each differentiate into CD8α /CD103 cDCs are short-lived, and their homeostatic maintenance relies + cDC1s and CD11b cDC2s, respectively. Although both cDCs rely on on constant replenishment from the BM precursors (5). The cy- the cytokine FLT3L during development, mechanisms that ensure tokine Fms-related tyrosine kinase 3 ligand (FLT3L) (12), by cDC accessibility to FLT3L have yet to be elucidated. Here, we gen- signaling through its receptor FLT3 expressed on DC precursors, erated mice that lacked a disintegrin and metalloproteinase (ADAM) is essential during the development of DCs (7, 13). -
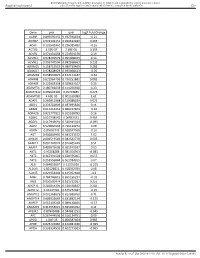
Gene Pval Qval Log2 Fold Change AAMP 0.895690332 0.952598834
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) Gut Gene pval qval Log2 Fold Change AAMP 0.895690332 0.952598834 -0.21 ABI3BP 0.002302151 0.020612283 0.465 ACHE 0.103542461 0.296385483 -0.16 ACTG2 2.99E-07 7.68E-05 3.195 ACVR1 0.071431098 0.224504378 0.19 ACVR1C 0.978209579 0.995008423 0.14 ACVRL1 0.006747504 0.042938663 0.235 ADAM15 0.158715519 0.380719469 0.285 ADAM17 0.978208929 0.995008423 -0.05 ADAM28 0.038932876 0.152174187 -0.62 ADAM8 0.622964796 0.790251882 0.085 ADAM9 0.122003358 0.329623107 0.25 ADAMTS1 0.180766659 0.414256926 0.23 ADAMTS12 0.009902195 0.05703885 0.425 ADAMTS8 4.60E-05 0.001169089 1.61 ADAP1 0.269811968 0.519388039 0.075 ADD1 0.233702809 0.487695826 0.11 ADM2 0.012213453 0.066227879 -0.36 ADRA2B 0.822777921 0.915518785 0.16 AEBP1 0.010738542 0.06035531 0.465 AGGF1 0.117946691 0.320915024 -0.095 AGR2 0.529860903 0.736120272 0.08 AGRN 0.85693743 0.928047568 -0.16 AGT 0.006849995 0.043233572 1.02 AHNAK 0.006519543 0.042542779 0.605 AKAP12 0.001747074 0.016405449 0.51 AKAP2 0.409929603 0.665919397 0.05 AKT1 0.95208288 0.985354963 -0.085 AKT2 0.367391504 0.620376005 0.055 AKT3 0.253556844 0.501934205 0.07 ALB 0.064833867 0.21195036 -0.315 ALDOA 0.83128831 0.918352939 0.08 ALOX5 0.029954404 0.125352668 -0.3 AMH 0.784746815 0.895196237 -0.03 ANG 0.050500474 0.181732067 0.255 ANGPT1 0.281853305 0.538528647 0.285 ANGPT2 0.43147281 0.675272487 -0.15 ANGPTL2 0.001368876 0.013688762 0.71 ANGPTL4 0.686032669 0.831882134 -0.175 ANPEP 0.019103243 0.089148466 -0.57 ANXA2P2 0.412553021 0.665966092 0.11 AP1M2 0.87843088 0.944681253 -0.045 APC 0.267444505 0.516134751 0.09 APOD 1.04E-05 0.000587404 0.985 APOE 0.023722987 0.104981036 -0.395 APOH 0.336334555 0.602273505 -0.065 Sundar R, et al. -

Inhibition of DDR1-BCR Signalling by Nilotinib As a New Therapeutic
Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer Maya Jeitany, Cédric Leroy, Priscillia Tosti, Marie Lafitte, Jordy Le Guet, Valérie Simon, Debora Bonenfant, Bruno Robert, Fanny Grillet, Caroline Mollevi, et al. To cite this version: Maya Jeitany, Cédric Leroy, Priscillia Tosti, Marie Lafitte, Jordy Le Guet, et al.. Inhibition of DDR1- BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer. EMBO Molecular Medicine, Wiley Open Access, 2018, 10 (4), pp.e7918. 10.15252/emmm.201707918. hal- 01872978 HAL Id: hal-01872978 https://hal.archives-ouvertes.fr/hal-01872978 Submitted on 12 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Research Article Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer Maya Jeitany1,†, Cédric Leroy1,2,3,†, Priscillia Tosti1,†, Marie Lafitte1, Jordy Le Guet1, Valérie Simon1, Debora Bonenfant2, Bruno Robert4, Fanny Grillet5, Caroline Mollevi4, Safia El Messaoudi4, Amaëlle Otandault4, Lucile Canterel-Thouennon4, Muriel Busson4, Alain R Thierry4, Pierre Martineau4, Julie Pannequin5, Serge Roche1,*,† & Audrey Sirvent1,†,** Abstract The current clinical management involves surgical removal of the primary tumour, often associated with chemotherapy. -

R&D Assay for Alzheimer's Disease
R&DR&D assayassay forfor Alzheimer’sAlzheimer’s diseasedisease Target screening⳼ Ⲽ㬔 antibody array, ᢜ⭉㬔 ⸽ἐⴐ Amyloid β-peptide Alzheimer’s disease⯸ ኸᷠ᧔ ᆹ⸽ inhibitor, antibody, ELISA kit Surwhrph#Surilohu#Dqwlerg|#Duud| 6OUSFBUFE 1."5SFBUFE )41 $3&# &3, &3, )41 $3&# &3, &3, 壤伡庰䋸TBNQMF ɅH 侴䋸嵄䍴䋸BOBMZUFT䋸䬱娴哜塵 1$ 1$ 1$ 1$ 5IFNPTUSFGFSFODFEBSSBZT 1$ 1$ QQ α 34, .4, 503 Q α 34, .4, 503 %SVHTDSFFOJOH0òUBSHFUFòFDUT0ATHWAY涭廐 6OUSFBUFE 堄币䋸4BNQMF侴䋸8FTUFSOPS&-*4"䍘䧽 1."5SFBUFE P 8FTUFSOCMPU廽喜儤应侴䋸0, Z 4VCTUSBUF -JHIU )31DPOKVHBUFE1BO "OUJQIPTQIPUZSPTJOF .FBO1JYFM%FOTJUZ Y $BQUVSF"OUJCPEZ 5BSHFU"OBMZUF "SSBZ.FNCSBOF $3&# &3, &3, )41 .4, Q α 34, 503 Human XL Cytokine Array kit (ARY022, 102 analytes) Adiponectin,Aggrecan,Angiogenin,Angiopoietin-1,Angiopoietin-2,BAFF,BDNF,Complement,Component C5/C5a,CD14,CD30,CD40L, Chitinase 3-like 1,Complement Factor D,C-Reactive Protein,Cripto-1,Cystatin C,Dkk-1,DPPIV,EGF,EMMPRIN,ENA-78,Endoglin, Fas L,FGF basic,FGF- 7,FGF-19,Flt-3 L,G-CSF,GDF-15,GM-CSF,GRO-α,Grow th Hormone,HGF,ICAM-1,IFN-γ,IGFBP-2,IGFBP-3, IL-1α,IL-1β, IL-1ra,IL-2,IL-3,IL-4,IL- 5,IL-6,IL-8, IL-10,IL-11,IL-12, IL-13,IL-15,IL-16,IL-17A,IL-18 BPa,IL-19,IL-22, IL-23,IL-24,IL-27, IL-31,IL-32α/β/γ,IL-33,IL-34,IP-10,I-TAC,Kallikrein 3,Leptin,LIF,Lipocalin-2,MCP-1,MCP-3,M-CSF,MIF,MIG,MIP-1α/MIP-1β,MIP-3α,MIP-3β,MMP-9, Myeloperoxidase,Osteopontin, p70, PDGF-AA, PDGF-AB/BB,Pentraxin-3, PF4, RAGE, RANTES,RBP4,Relaxin-2, Resistin,SDF-1α,Serpin E1, SHBG, ST2, TARC,TFF3,TfR,TGF- ,Thrombospondin-1,TNF-α, uPAR, VEGF, Vitamin D BP Human Protease (34 analytes) / -

60+ Genes Tested FDA-Approved Targeted Therapies & Gene Indicators
® 60+ genes tested ABL1, ABL2, ALK, AR, ARAF, ATM, ATR, BRAF, BRCA1*, BRCA2*, BTK, CCND1, CCND2, CCND3, CDK4, Somatic mutation detection CDK6, CDKN1A, CDKN1B, CDKN2A, CDKN2B, DDR1, DDR2, EGFR, ERBB2 (HER2), ESR1, FGFR1, FGFR2, for approved cancer therapies FGFR3, FGFR4, FLCN, FLT1, FLT3, FLT4, GNA11, GNAQ, HDAC1, HDAC2, HRAS, JAK1, JAK2, KDR, KIT, in solid tumors KRAS, MAP2K1, MET, MTOR, NF1, NF2, NRAS, PALB2, PARP1, PDGFRA, PDGFRB, PIK3CA, PIK3CD, PTCH1, PTEN, RAF1, RET, ROS1, SMO, SRC, STK11, TNK2, TSC1, TSC2 FDA-approved Targeted Therapies & Gene Indicators Abiraterone AR Necitumumab EGFR Ado-Trastuzumab ERBB2 (HER2) Nilotinib ABL1, ABL2, DDR1, DDR2, KIT, PDGFRA, PDGFRB Emtansine FGFR1, FGFR2, FGFR3, FLT1, FLT4, KDR, PDGFRA, Afatinib EGFR, ERBB2 (HER2) Nintedanib PDGFRB Alectinib ALK Olaparib ATM, ATR, BRCA1*, BRCA2*, PALB2, PARP1 Anastrozole ESR1 Osimertinib EGFR Axitinib FLT1, FLT4, KDR, KIT, PDGFRA, PDGFRB CDK4, CDK6, CCND1, CCND2, CCND3, CDKN1A, Palbociclib Belinostat HDAC1, HDAC2 CDKN1B, CDKN2A, CDKN2B Panitumumab EGFR Bicalutamide AR Panobinostat HDAC1, HDAC2 Bosutinib ABL1, SRC Pazopanib FLT1, FLT4, KDR, KIT, PDGFRA, PDGFRB Cabozantinib FLT1, FLT3, FLT4, KDR, KIT, MET, RET Pertuzumab ERBB2 (HER2) Ceritinib ALK ABL1, FGFR1, FGFR2, FGFR3, FGFR4, FLT1, FLT3, FLT4, Cetuximab EGFR Ponatinib KDR, KIT, PDGFRA, PDGFRB, RET, SRC Cobimetinib MAP2K1 Ramucirumab KDR Crizotinib ALK, MET, ROS1 Regorafenib ARAF, BRAF, FLT1, FLT4, KDR, KIT, PDGFRB, RAF1, RET Dabrafenib BRAF Ruxolitinib JAK1, JAK2 Dasatinib ABL1, ABL2, DDR1, DDR2, SRC, TNK2 -

CD Markers Are Routinely Used for the Immunophenotyping of Cells
ptglab.com 1 CD MARKER ANTIBODIES www.ptglab.com Introduction The cluster of differentiation (abbreviated as CD) is a protocol used for the identification and investigation of cell surface molecules. So-called CD markers are routinely used for the immunophenotyping of cells. Despite this use, they are not limited to roles in the immune system and perform a variety of roles in cell differentiation, adhesion, migration, blood clotting, gamete fertilization, amino acid transport and apoptosis, among many others. As such, Proteintech’s mini catalog featuring its antibodies targeting CD markers is applicable to a wide range of research disciplines. PRODUCT FOCUS PECAM1 Platelet endothelial cell adhesion of blood vessels – making up a large portion molecule-1 (PECAM1), also known as cluster of its intracellular junctions. PECAM-1 is also CD Number of differentiation 31 (CD31), is a member of present on the surface of hematopoietic the immunoglobulin gene superfamily of cell cells and immune cells including platelets, CD31 adhesion molecules. It is highly expressed monocytes, neutrophils, natural killer cells, on the surface of the endothelium – the thin megakaryocytes and some types of T-cell. Catalog Number layer of endothelial cells lining the interior 11256-1-AP Type Rabbit Polyclonal Applications ELISA, FC, IF, IHC, IP, WB 16 Publications Immunohistochemical of paraffin-embedded Figure 1: Immunofluorescence staining human hepatocirrhosis using PECAM1, CD31 of PECAM1 (11256-1-AP), Alexa 488 goat antibody (11265-1-AP) at a dilution of 1:50 anti-rabbit (green), and smooth muscle KD/KO Validated (40x objective). alpha-actin (red), courtesy of Nicola Smart. PECAM1: Customer Testimonial Nicola Smart, a cardiovascular researcher “As you can see [the immunostaining] is and a group leader at the University of extremely clean and specific [and] displays Oxford, has said of the PECAM1 antibody strong intercellular junction expression, (11265-1-AP) that it “worked beautifully as expected for a cell adhesion molecule.” on every occasion I’ve tried it.” Proteintech thanks Dr. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal