Identification of Selection Signatures Involved In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Long-Read Genome Sequencing for the Diagnosis Of
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.02.185447; this version posted September 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Long-read genome sequencing for the diagnosis of neurodevelopmental disorders Susan M. Hiatt1, James M.J. Lawlor1, Lori H. Handley1, Ryne C. Ramaker1, Brianne B. Rogers1,2, E. Christopher Partridge1, Lori Beth Boston1, Melissa Williams1, Christopher B. Plott1, Jerry Jenkins1, David E. Gray1, James M. Holt1, Kevin M. Bowling1, E. Martina Bebin3, Jane Grimwood1, Jeremy Schmutz1, Gregory M. Cooper1* 1HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA, 35806 2Department of Genetics, University of Alabama at Birmingham, Birmingham, AL, USA, 35924 3Department of Neurology, University of Alabama at Birmingham, Birmingham, AL, USA, 35924 *[email protected], 256-327-9490 Conflicts of Interest The authors all declare no conflicts of interest. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.02.185447; this version posted September 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Purpose Exome and genome sequencing have proven to be effective tools for the diagnosis of neurodevelopmental disorders (NDDs), but large fractions of NDDs cannot be attributed to currently detectable genetic variation. This is likely, at least in part, a result of the fact that many genetic variants are difficult or impossible to detect through typical short-read sequencing approaches. -

Mouse Ppef1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ppef1 Knockout Project (CRISPR/Cas9) Objective: To create a Ppef1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ppef1 gene (NCBI Reference Sequence: NM_011147 ; Ensembl: ENSMUSG00000062168 ) is located on Mouse chromosome X. 16 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 16 (Transcript: ENSMUST00000071204). Exon 2~4 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygous null female and hemizygous null male mice are viable, fertile and display no overt abnormalities. Exon 2 starts from about 2.41% of the coding region. Exon 2~4 covers 18.41% of the coding region. The size of effective KO region: ~10105 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 16 Legends Exon of mouse Ppef1 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 4 is aligned with itself to determine if there are tandem repeats. -

Identification of Conserved Genes Triggering Puberty in European Sea
Blázquez et al. BMC Genomics (2017) 18:441 DOI 10.1186/s12864-017-3823-2 RESEARCHARTICLE Open Access Identification of conserved genes triggering puberty in European sea bass males (Dicentrarchus labrax) by microarray expression profiling Mercedes Blázquez1,2* , Paula Medina1,2,3, Berta Crespo1,4, Ana Gómez1 and Silvia Zanuy1* Abstract Background: Spermatogenesisisacomplexprocesscharacterized by the activation and/or repression of a number of genes in a spatio-temporal manner. Pubertal development in males starts with the onset of the first spermatogenesis and implies the division of primary spermatogonia and their subsequent entry into meiosis. This study is aimed at the characterization of genes involved in the onset of puberty in European sea bass, and constitutes the first transcriptomic approach focused on meiosis in this species. Results: European sea bass testes collected at the onset of puberty (first successful reproduction) were grouped in stage I (resting stage), and stage II (proliferative stage). Transition from stage I to stage II was marked by an increase of 11ketotestosterone (11KT), the main fish androgen, whereas the transcriptomic study resulted in 315 genes differentially expressed between the two stages. The onset of puberty induced 1) an up-regulation of genes involved in cell proliferation, cell cycle and meiosis progression, 2) changes in genes related with reproduction and growth, and 3) a down-regulation of genes included in the retinoic acid (RA) signalling pathway. The analysis of GO-terms and biological pathways showed that cell cycle, cell division, cellular metabolic processes, and reproduction were affected, consistent with the early events that occur during the onset of puberty. -

PPEF1 Monoclonal Antibody (M01), Clone 1F6-1A5
PPEF1 monoclonal antibody (M01), clone 1F6-1A5 Catalog # : H00005475-M01 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a full length recombinant Western Blot (Transfected lysate) Description: PPEF1. Immunogen: PPEF1 (AAH36026, 1 a.a. ~ 653 a.a) full-length recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: MGCSSSSTKTRRSDTSLRAALIIQNWYRGYKARLKARQHYALTIFQSIEY ADEQGQMQLSTFFSFMLENYTHIHKEELELRNQSLESEQDMRDRWDYV DSIDVPDSYNGPRLQFPLTCTDIDLLLEAFKEQQILHAHYVLEVLFETKKV enlarge LKQMPNFTHIQTSPSKEVTICGDLHGKLDDLFLIFYKNGLPSERNPYVFNG Western Blot (Recombinant DFVDRGKNSIEILMILCVSFLVYPNDLHLNRGNHEDFMMNLRYGFTKEILH protein) KYKLHGKRILQILEEFYAWLPIGTIVDNEILVIHGGISETTDLNLLHRVERNK MKSVLIPPTETNRDHDTDSKHNKVGVTFNAHGRIKTNGSPTEHLTEHEWE ELISA QIIDILWSDPRGKNGCFPNTCRGGGCYFGPDVTSKILNKYQLKMLIRSHE CKPEGYEICHDGKVVTIFSASNYYEEGSNRGAYIKLCSGTTPRFFQYQVT RNAi Knockdown (Antibody KATCFQPLRQRVDTMENSAIKILRERVISRKSDLTRAFQLQDHRKSGKLS validated) VSQWAFCMENILGLNLPWRSLSSNLVNIDQNGNVEYMSSFQNIRIEKPVQ EAHSTLVETLYRYRSDLEIIFNAIDTDHSGLISVEEFRAMWKLFSSHYNVHI DDSQVNKLANIMDLNKDGSIDFNEFLKAFYVVHRYEDLMKPDVTNLG Host: Mouse Reactivity: Human enlarge Isotype: IgG1 Kappa Quality Control Antibody Reactive Against Recombinant Protein. Testing: Western Blot detection against Immunogen (97.57 KDa) . Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download Datasheet: Download Applications Page 1 of 3 2016/5/21 Western Blot (Transfected lysate) -

Frequent Loss of Heterozygosity Targeting the Inactive X Chromosome in Melanoma
6476 Vol. 9, 6476–6482, December 15, 2003 Clinical Cancer Research Frequent Loss of Heterozygosity Targeting the Inactive X Chromosome in Melanoma James O. Indsto,1 Najah T. Nassif,2 INTRODUCTION Richard F. Kefford,1 and Graham J. Mann1 Chromosomal deletions and amplification events are fre- 1Westmead Institute for Cancer Research, University of Sydney at quent in advanced neoplasms and provide evidence for the Westmead Millennium Institute, Westmead, New South Wales, existence and location of putative tumor suppressor genes Australia, and 2Department of Cell and Molecular Biology, University (TSGs) and oncogenes. In melanoma, 9p and 10q deletions are of Technology, Sydney, New South Wales, Australia most frequent, and losses on 6q, 11q, 1p, 15p, 17q, and 18q are also frequently found (1, 2). However, no studies have focused on the X chromosome. In the human female, random inactiva- ABSTRACT tion of one copy of an X chromosome by hypermethylation is an After previous preliminary observations of paradoxical early event in embryogenesis, resulting in most tissues being a deletion events affecting the inactive X chromosome in mel- mosaic in terms of X inactivation status. Neoplasms, being anoma, we have surveyed the X chromosome for deletions clonal expansions of single precursor cells, usually share a using 23 polymorphic microsatellite markers in 28 inform- common X inactivation status. This can be conveniently assayed ative (female XX) metastatic melanomas. Ten tumors (36%) using the methylation-sensitive restriction enzyme HpaII, which showed at least one loss of heterozygosity (LOH) event, and has a recognition site within the highly polymorphic exon 1 in two cases an entire chromosome showed LOH at all CAG repeat (ARTR) in the androgen receptor (AR) gene. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root
Research Paper Comparative transcriptome profiling of the human and mouse dorsal root ganglia: an RNA-seq–based resource for pain and sensory neuroscience research a,b a a a a a 07/20/2018 on BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3mH5nK33R3Qh4f27oe7zFUUf7ZAUK5aCsyqAeT54jiDxP7ZjumT3TrA== by https://journals.lww.com/pain from Downloaded Pradipta Ray , Andrew Torck , Lilyana Quigley , Andi Wangzhou , Matthew Neiman , Chandranshu Rao , Downloaded Tiffany Lama, Ji-Young Kima, Tae Hoon Kimb, Michael Q. Zhangb, Gregory Dussora, Theodore J. Pricea,* from https://journals.lww.com/pain Abstract Molecular neurobiological insight into human nervous tissues is needed to generate next-generation therapeutics for neurological disorders such as chronic pain. We obtained human dorsal root ganglia (hDRG) samples from organ donors and performed RNA- sequencing (RNA-seq) to study the hDRG transcriptional landscape, systematically comparing it with publicly available data from by BhDMf5ePHKav1zEoum1tQfN4a+kJLhEZgbsIHo4XMi0hCywCX1AWnYQp/IlQrHD3mH5nK33R3Qh4f27oe7zFUUf7ZAUK5aCsyqAeT54jiDxP7ZjumT3TrA== a variety of human and orthologous mouse tissues, including mouse DRG (mDRG). We characterized the hDRG transcriptional profile in terms of tissue-restricted gene coexpression patterns and putative transcriptional regulators, and formulated an information-theoretic framework to quantify DRG enrichment. Relevant gene families and pathways were also analyzed, including transcription factors, G-protein-coupled receptors, and ion channels. Our analyses reveal an hDRG-enriched protein-coding gene set (;140), some of which have not been described in the context of DRG or pain signaling. Most of these show conserved enrichment in mDRG and were mined for known drug–gene product interactions. Conserved enrichment of the vast majority of transcription factors suggests that the mDRG is a faithful model system for studying hDRG, because of evolutionarily conserved regulatory programs. -

Clinically Annotated Breast, Ovarian and Pancreatic Cancer
www.nature.com/scientificreports OPEN MetaGxData: Clinically Annotated Breast, Ovarian and Pancreatic Cancer Datasets and their Use in Received: 19 November 2018 Accepted: 31 May 2019 Generating a Multi-Cancer Gene Published: xx xx xxxx Signature Deena M. A. Gendoo 1, Michael Zon2,4, Vandana Sandhu2, Venkata S. K. Manem2,3,5, Natchar Ratanasirigulchai2, Gregory M. Chen2, Levi Waldron 6 & Benjamin Haibe- Kains2,3,7,8,9 A wealth of transcriptomic and clinical data on solid tumours are under-utilized due to unharmonized data storage and format. We have developed the MetaGxData package compendium, which includes manually-curated and standardized clinical, pathological, survival, and treatment metadata across breast, ovarian, and pancreatic cancer data. MetaGxData is the largest compendium of curated transcriptomic data for these cancer types to date, spanning 86 datasets and encompassing 15,249 samples. Open access to standardized metadata across cancer types promotes use of their transcriptomic and clinical data in a variety of cross-tumour analyses, including identifcation of common biomarkers, and assessing the validity of prognostic signatures. Here, we demonstrate that MetaGxData is a fexible framework that facilitates meta-analyses by using it to identify common prognostic genes in ovarian and breast cancer. Furthermore, we use the data compendium to create the frst gene signature that is prognostic in a meta-analysis across 3 cancer types. These fndings demonstrate the potential of MetaGxData to serve as an important resource in oncology research, and provide a foundation for future development of cancer-specifc compendia. Ovarian, breast and pancreatic cancers are among the leading causes of cancer deaths among women, and recent studies have identifed biological and molecular commonalities between them1–4. -

Data-Driven Analysis of Age, Sex, and Tissue Effects on Gene Expression Variability in Alzheimer’S Disease Lavida R.K
bioRxiv preprint doi: https://doi.org/10.1101/498527; this version posted December 17, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Data-Driven Analysis of Age, Sex, and Tissue Effects on Gene Expression Variability in Alzheimer’s Disease Lavida R.K. Brooks 1, George I. Mias,2;∗ 1 Institute for Quantitative Health Science and Engineering, Michigan State University, Microbiology and Molecular Genetics, East Lansing, 48824, USA 2 Institute for Quantitative Health Science and Engineering, Michigan State University, Biochemistry and Molecular Biology, East Lansing, 48824, USA Correspondence*: George I. Mias [email protected] 2 ABSTRACT 3 Alzheimer’s disease (AD) has been categorized by the Centers for Disease Control and 4 Prevention (CDC) as the 6th leading cause of death in the United States. AD is a significant 5 health-care burden because of its increased occurrence (specifically in the elderly population) 6 and the lack of effective treatments and preventive methods. With an increase in life expectancy, 7 the CDC expects AD cases to rise to 15 million by 2060. Aging has been previously associated 8 with susceptibility to AD, and there are ongoing efforts to effectively differentiate between normal 9 and AD age-related brain degeneration and memory loss. AD targets neuronal function and can 10 cause neuronal loss due to the buildup of amyloid-beta plaques and intracellular neurofibrillary 11 tangles. 12 Our study aims to identify temporal changes within gene expression profiles of healthy controls 13 and AD subjects. -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -
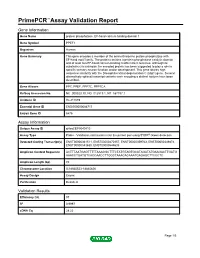
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name protein phosphatase, EF-hand calcium binding domain 1 Gene Symbol PPEF1 Organism Human Gene Summary This gene encodes a member of the serine/threonine protein phosphatase with EF-hand motif family. The protein contains a protein phosphatase catalytic domain and at least two EF-hand calcium-binding motifs in its C terminus. Although its substrate(s) is unknown the encoded protein has been suggested to play a role in specific sensory neuron function and/or development. This gene shares high sequence similarity with the Drosophila retinal degeneration C (rdgC) gene. Several alternatively spliced transcript variants each encoding a distinct isoform have been described. Gene Aliases PP7, PPEF, PPP7C, PPP7CA RefSeq Accession No. NC_000023.10, NG_012519.1, NT_167197.1 UniGene ID Hs.211589 Ensembl Gene ID ENSG00000086717 Entrez Gene ID 5475 Assay Information Unique Assay ID qHsaCEP0049810 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000361511, ENST00000470157, ENST00000359763, ENST00000349874, ENST00000543630, ENST00000544635 Amplicon Context Sequence ACTTTAATGAGTTTTTAAAGGCTTTCTATGTAGTGCATAGATATGAAGACTTGATG AAACCTGATGTCACCAACCTTGGCTAAACACAAATGAGAGCTTCCCTC Amplicon Length (bp) 74 Chromosome Location X:18845523-18845626 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9997 cDNA Cq 28.22 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 78 gDNA Cq 25.76 Specificity -

Functional Genomics Approach Identifies Novel Signaling Regulators of Tgfa Ectodomain Shedding
Published OnlineFirst October 10, 2017; DOI: 10.1158/1541-7786.MCR-17-0140 Signal Transduction Molecular Cancer Research Functional Genomics Approach Identifies Novel Signaling Regulators of TGFa Ectodomain Shedding Jennifer L. Wilson1, Eirini Kefaloyianni2, Lauren Stopfer1, Christina Harrison3, Venkata S. Sabbisetti4, Ernest Fraenkel1,5, Douglas A. Lauffenburger1, and Andreas Herrlich2 Abstract Ectodomain shedding of cell-surface precursor proteins by shRNA genomic screen, computational network analysis, and metalloproteases generates important cellular signaling molecules. dedicated validation tests succeeded in identifying several key Of importance for disease is the release of ligands that activate the signaling pathways as novel regulators of TGFa shedding in cancer EGFR, such as TGFa, which is mostly carried out by ADAM17 [a cells. Most notably, a cluster of genes with NFkBpathwayregu- member of the A-disintegrin and metalloprotease (ADAM) latory functions was found to strongly influence TGFa release, domain family]. EGFR ligand shedding has been linked to many albeit independent of their NFkB regulatory functions. Inflamma- diseases, in particular cancer development, growth and metastasis, tory regulators thus also govern cancer cell growth–promoting as well as resistance to cancer therapeutics. Excessive EGFR ligand ectodomain cleavage, lending mechanistic understanding to the release can outcompete therapeutic EGFR inhibition or the inhi- well-known connection between inflammation and cancer. bition of other growth factor pathways by providing bypass signaling via EGFR activation. Drugging metalloproteases directly Implications: Using genomic screens and network analysis, this have failed clinically because it indiscriminately affected shedding study defines targets that regulate ectodomain shedding and of numerous substrates. It is therefore essential to identify regu- suggests new treatment opportunities for EGFR-driven cancers.