Data-Driven Analysis of Age, Sex, and Tissue Effects on Gene Expression Variability in Alzheimer’S Disease Lavida R.K
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 Long-Read Genome Sequencing for the Diagnosis Of
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.02.185447; this version posted September 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Long-read genome sequencing for the diagnosis of neurodevelopmental disorders Susan M. Hiatt1, James M.J. Lawlor1, Lori H. Handley1, Ryne C. Ramaker1, Brianne B. Rogers1,2, E. Christopher Partridge1, Lori Beth Boston1, Melissa Williams1, Christopher B. Plott1, Jerry Jenkins1, David E. Gray1, James M. Holt1, Kevin M. Bowling1, E. Martina Bebin3, Jane Grimwood1, Jeremy Schmutz1, Gregory M. Cooper1* 1HudsonAlpha Institute for Biotechnology, Huntsville, AL, USA, 35806 2Department of Genetics, University of Alabama at Birmingham, Birmingham, AL, USA, 35924 3Department of Neurology, University of Alabama at Birmingham, Birmingham, AL, USA, 35924 *[email protected], 256-327-9490 Conflicts of Interest The authors all declare no conflicts of interest. 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.02.185447; this version posted September 14, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract Purpose Exome and genome sequencing have proven to be effective tools for the diagnosis of neurodevelopmental disorders (NDDs), but large fractions of NDDs cannot be attributed to currently detectable genetic variation. This is likely, at least in part, a result of the fact that many genetic variants are difficult or impossible to detect through typical short-read sequencing approaches. -

Mouse Ppef1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ppef1 Knockout Project (CRISPR/Cas9) Objective: To create a Ppef1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ppef1 gene (NCBI Reference Sequence: NM_011147 ; Ensembl: ENSMUSG00000062168 ) is located on Mouse chromosome X. 16 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 16 (Transcript: ENSMUST00000071204). Exon 2~4 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygous null female and hemizygous null male mice are viable, fertile and display no overt abnormalities. Exon 2 starts from about 2.41% of the coding region. Exon 2~4 covers 18.41% of the coding region. The size of effective KO region: ~10105 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 16 Legends Exon of mouse Ppef1 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of Exon 2 is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of Exon 4 is aligned with itself to determine if there are tandem repeats. -

Identification of Conserved Genes Triggering Puberty in European Sea
Blázquez et al. BMC Genomics (2017) 18:441 DOI 10.1186/s12864-017-3823-2 RESEARCHARTICLE Open Access Identification of conserved genes triggering puberty in European sea bass males (Dicentrarchus labrax) by microarray expression profiling Mercedes Blázquez1,2* , Paula Medina1,2,3, Berta Crespo1,4, Ana Gómez1 and Silvia Zanuy1* Abstract Background: Spermatogenesisisacomplexprocesscharacterized by the activation and/or repression of a number of genes in a spatio-temporal manner. Pubertal development in males starts with the onset of the first spermatogenesis and implies the division of primary spermatogonia and their subsequent entry into meiosis. This study is aimed at the characterization of genes involved in the onset of puberty in European sea bass, and constitutes the first transcriptomic approach focused on meiosis in this species. Results: European sea bass testes collected at the onset of puberty (first successful reproduction) were grouped in stage I (resting stage), and stage II (proliferative stage). Transition from stage I to stage II was marked by an increase of 11ketotestosterone (11KT), the main fish androgen, whereas the transcriptomic study resulted in 315 genes differentially expressed between the two stages. The onset of puberty induced 1) an up-regulation of genes involved in cell proliferation, cell cycle and meiosis progression, 2) changes in genes related with reproduction and growth, and 3) a down-regulation of genes included in the retinoic acid (RA) signalling pathway. The analysis of GO-terms and biological pathways showed that cell cycle, cell division, cellular metabolic processes, and reproduction were affected, consistent with the early events that occur during the onset of puberty. -

PPEF1 Monoclonal Antibody (M01), Clone 1F6-1A5
PPEF1 monoclonal antibody (M01), clone 1F6-1A5 Catalog # : H00005475-M01 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a full length recombinant Western Blot (Transfected lysate) Description: PPEF1. Immunogen: PPEF1 (AAH36026, 1 a.a. ~ 653 a.a) full-length recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: MGCSSSSTKTRRSDTSLRAALIIQNWYRGYKARLKARQHYALTIFQSIEY ADEQGQMQLSTFFSFMLENYTHIHKEELELRNQSLESEQDMRDRWDYV DSIDVPDSYNGPRLQFPLTCTDIDLLLEAFKEQQILHAHYVLEVLFETKKV enlarge LKQMPNFTHIQTSPSKEVTICGDLHGKLDDLFLIFYKNGLPSERNPYVFNG Western Blot (Recombinant DFVDRGKNSIEILMILCVSFLVYPNDLHLNRGNHEDFMMNLRYGFTKEILH protein) KYKLHGKRILQILEEFYAWLPIGTIVDNEILVIHGGISETTDLNLLHRVERNK MKSVLIPPTETNRDHDTDSKHNKVGVTFNAHGRIKTNGSPTEHLTEHEWE ELISA QIIDILWSDPRGKNGCFPNTCRGGGCYFGPDVTSKILNKYQLKMLIRSHE CKPEGYEICHDGKVVTIFSASNYYEEGSNRGAYIKLCSGTTPRFFQYQVT RNAi Knockdown (Antibody KATCFQPLRQRVDTMENSAIKILRERVISRKSDLTRAFQLQDHRKSGKLS validated) VSQWAFCMENILGLNLPWRSLSSNLVNIDQNGNVEYMSSFQNIRIEKPVQ EAHSTLVETLYRYRSDLEIIFNAIDTDHSGLISVEEFRAMWKLFSSHYNVHI DDSQVNKLANIMDLNKDGSIDFNEFLKAFYVVHRYEDLMKPDVTNLG Host: Mouse Reactivity: Human enlarge Isotype: IgG1 Kappa Quality Control Antibody Reactive Against Recombinant Protein. Testing: Western Blot detection against Immunogen (97.57 KDa) . Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download Datasheet: Download Applications Page 1 of 3 2016/5/21 Western Blot (Transfected lysate) -

Frequent Loss of Heterozygosity Targeting the Inactive X Chromosome in Melanoma
6476 Vol. 9, 6476–6482, December 15, 2003 Clinical Cancer Research Frequent Loss of Heterozygosity Targeting the Inactive X Chromosome in Melanoma James O. Indsto,1 Najah T. Nassif,2 INTRODUCTION Richard F. Kefford,1 and Graham J. Mann1 Chromosomal deletions and amplification events are fre- 1Westmead Institute for Cancer Research, University of Sydney at quent in advanced neoplasms and provide evidence for the Westmead Millennium Institute, Westmead, New South Wales, existence and location of putative tumor suppressor genes Australia, and 2Department of Cell and Molecular Biology, University (TSGs) and oncogenes. In melanoma, 9p and 10q deletions are of Technology, Sydney, New South Wales, Australia most frequent, and losses on 6q, 11q, 1p, 15p, 17q, and 18q are also frequently found (1, 2). However, no studies have focused on the X chromosome. In the human female, random inactiva- ABSTRACT tion of one copy of an X chromosome by hypermethylation is an After previous preliminary observations of paradoxical early event in embryogenesis, resulting in most tissues being a deletion events affecting the inactive X chromosome in mel- mosaic in terms of X inactivation status. Neoplasms, being anoma, we have surveyed the X chromosome for deletions clonal expansions of single precursor cells, usually share a using 23 polymorphic microsatellite markers in 28 inform- common X inactivation status. This can be conveniently assayed ative (female XX) metastatic melanomas. Ten tumors (36%) using the methylation-sensitive restriction enzyme HpaII, which showed at least one loss of heterozygosity (LOH) event, and has a recognition site within the highly polymorphic exon 1 in two cases an entire chromosome showed LOH at all CAG repeat (ARTR) in the androgen receptor (AR) gene. -

Translational Control in Cancer Etiology
Downloaded from http://cshperspectives.cshlp.org/ on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Translational Control in Cancer Etiology Davide Ruggero Helen Diller Cancer Center, School of Medicine, University of California, San Francisco, California 94158 Correspondence: [email protected] The link between perturbations in translational control and cancer etiology is becoming a primary focus in cancer research. It has now been established that genetic alterations in several components of the translational apparatus underlie spontaneous cancers as well as an entire class of inherited syndromes known as “ribosomopathies” associated with in- creased cancer susceptibility. These discoveries have illuminated the importance of dereg- ulations in translational control to very specific cellular processes that contribute to cancer etiology. In addition, a growing body of evidence supports the view that deregulation of translational control is a common mechanism by which diverse oncogenic pathways promote cellular transformation and tumor development. Indeed, activation of these key oncogenic pathways induces rapid and dramatic translational reprogramming both by in- creasing overall protein synthesis and by modulating specific mRNA networks. These trans- lational changes promote cellular transformation, impacting almost every phase of tumor development. This paradigm represents a new frontier in the multihit model of cancer for- mation and offers significant promise for innovative cancer therapies. Current research, -

Disease-Related Cellular Protein Networks Differentially Affected
www.nature.com/scientificreports OPEN Disease‑related cellular protein networks diferentially afected under diferent EGFR mutations in lung adenocarcinoma Toshihide Nishimura1,8*, Haruhiko Nakamura1,2,8, Ayako Yachie3,8, Takeshi Hase3,8, Kiyonaga Fujii1,8, Hirotaka Koizumi4, Saeko Naruki4, Masayuki Takagi4, Yukiko Matsuoka3, Naoki Furuya5, Harubumi Kato6,7 & Hisashi Saji2 It is unclear how epidermal growth factor receptor EGFR major driver mutations (L858R or Ex19del) afect downstream molecular networks and pathways. This study aimed to provide information on the infuences of these mutations. The study assessed 36 protein expression profles of lung adenocarcinoma (Ex19del, nine; L858R, nine; no Ex19del/L858R, 18). Weighted gene co-expression network analysis together with analysis of variance-based screening identifed 13 co-expressed modules and their eigen proteins. Pathway enrichment analysis for the Ex19del mutation demonstrated involvement of SUMOylation, epithelial and mesenchymal transition, ERK/mitogen- activated protein kinase signalling via phosphorylation and Hippo signalling. Additionally, analysis for the L858R mutation identifed various pathways related to cancer cell survival and death. With regard to the Ex19del mutation, ROCK, RPS6KA1, ARF1, IL2RA and several ErbB pathways were upregulated, whereas AURK and GSKIP were downregulated. With regard to the L858R mutation, RB1, TSC22D3 and DOCK1 were downregulated, whereas various networks, including VEGFA, were moderately upregulated. In all mutation types, CD80/CD86 (B7), MHC, CIITA and IFGN were activated, whereas CD37 and SAFB were inhibited. Costimulatory immune-checkpoint pathways by B7/CD28 were mainly activated, whereas those by PD-1/PD-L1 were inhibited. Our fndings may help identify potential therapeutic targets and develop therapeutic strategies to improve patient outcomes. -
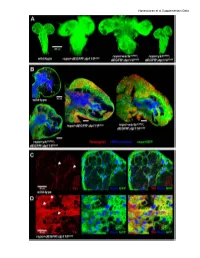
Supplementary Data Vigneswaran Et Al Supplementary Data
Vigneswaran et al Supplementary Data Vigneswaran et al Supplementary Data Figure S1: Yki is required for EGFR-PI3K-driven glial neoplasia in Drosophila (A) Optical projections of whole brain-nerve cord complexes from 3rd instar larvae approximately 130 hrs old. Dorsal view; anterior up. CD8-GFP (green) labels glial cell bodies. Compared to repo>dEGFRλ;dp110CAAX, warts knockdown (repo>wartsdsRNA; dEGFRλ;dp110CAAX) increased neoplastic brain overgrowth and yki knockdown (repo>ykidsRNA;dEGFRλ;dp110CAAX) decreased neoplastic brain overgrowth. (B) 3 µm optical projections of brain hemispheres, age-matched 3rd instar larvae. Frontal sections; anterior up; midline to left. Repo (red) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies; anti-HRP (blue) counter-stains for neurons and neuropil. (middle) repo>dEGFRλ;dp110CAAX showed increased glial cell numbers (red nuclei) compared to (upper left) wild-type. Compared to repo>dEGFRλ;dp110CAAX, (right) warts knockdown increased neoplastic glial cell numbers (red nuclei), whereas (lower left) yki knockdown reduced neoplastic glial cell numbers (red nuclei). (C, D) Low levels of Yki protein (red) was observed in wild-type central brain glia (white arrows, left panel in C) compared to high levels of cytoplasmic and nuclear Yki protein in dEGFRλ;dp110CAAX neoplastic glia (white arrows, left panel in D); Repo (blue) labels glial cell nuclei; CD8-GFP (green) labels glial cell bodies. Vigneswaran et al Supplementary Data Figure S2: YAP/TAZ expression confined to RTK-amplified tuMor cells and Maintained in patient-derived xenografts (A) On the left, immunohistochemical (IHC) staining in representative normal brain parenchyma in the cortex where YAP expression and TAZ expression was limited to vascular cells and was not detectable in normal neuronal and glial cells. -

A Master Autoantigen-Ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. A Master Autoantigen-ome Links Alternative Splicing, Female Predilection, and COVID-19 to Autoimmune Diseases Julia Y. Wang1*, Michael W. Roehrl1, Victor B. Roehrl1, and Michael H. Roehrl2* 1 Curandis, New York, USA 2 Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, USA * Correspondence: [email protected] or [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.30.454526; this version posted August 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Chronic and debilitating autoimmune sequelae pose a grave concern for the post-COVID-19 pandemic era. Based on our discovery that the glycosaminoglycan dermatan sulfate (DS) displays peculiar affinity to apoptotic cells and autoantigens (autoAgs) and that DS-autoAg complexes cooperatively stimulate autoreactive B1 cell responses, we compiled a database of 751 candidate autoAgs from six human cell types. At least 657 of these have been found to be affected by SARS-CoV-2 infection based on currently available multi-omic COVID data, and at least 400 are confirmed targets of autoantibodies in a wide array of autoimmune diseases and cancer. -

Comparative Transcriptome Profiling of the Human and Mouse Dorsal Root Ganglia: an RNA-Seq-Based Resource for Pain and Sensory Neuroscience Research
bioRxiv preprint doi: https://doi.org/10.1101/165431; this version posted October 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Title: Comparative transcriptome profiling of the human and mouse dorsal root ganglia: An RNA-seq-based resource for pain and sensory neuroscience research Short Title: Human and mouse DRG comparative transcriptomics Pradipta Ray 1, 2 #, Andrew Torck 1 , Lilyana Quigley 1, Andi Wangzhou 1, Matthew Neiman 1, Chandranshu Rao 1, Tiffany Lam 1, Ji-Young Kim 1, Tae Hoon Kim 2, Michael Q. Zhang 2, Gregory Dussor 1 and Theodore J. Price 1, # 1 The University of Texas at Dallas, School of Behavioral and Brain Sciences 2 The University of Texas at Dallas, Department of Biological Sciences # Corresponding authors Theodore J Price Pradipta Ray School of Behavioral and Brain Sciences School of Behavioral and Brain Sciences The University of Texas at Dallas The University of Texas at Dallas BSB 14.102G BSB 10.608 800 W Campbell Rd 800 W Campbell Rd Richardson TX 75080 Richardson TX 75080 972-883-4311 972-883-7262 [email protected] [email protected] Number of pages: 27 Number of figures: 9 Number of tables: 8 Supplementary Figures: 4 Supplementary Files: 6 Word count: Abstract = 219; Introduction = 457; Discussion = 1094 Conflict of interest: The authors declare no conflicts of interest Patient anonymity and informed consent: Informed consent for human tissue sources were obtained by Anabios, Inc. (San Diego, CA). Human studies: This work was approved by The University of Texas at Dallas Institutional Review Board (MR 15-237). -

Identification of Selection Signatures Involved In
Almeida et al. BMC Genomics (2019) 20:449 https://doi.org/10.1186/s12864-019-5811-1 RESEARCHARTICLE Open Access Identification of selection signatures involved in performance traits in a paternal broiler line Octávio Augusto Costa Almeida1, Gabriel Costa Monteiro Moreira1, Fernanda Marcondes Rezende2, Clarissa Boschiero3, Jane de Oliveira Peixoto4, Adriana Mercia Guaratini Ibelli4, Mônica Corrêa Ledur4, Francisco José de Novais1 and Luiz Lehmann Coutinho1* Abstract Background: Natural and artificial selection leads to changes in certain regions of the genome resulting in selection signatures that can reveal genes associated with the selected traits. Selection signatures may be identified using different methodologies, of which some are based on detecting contiguous sequences of homozygous identical-by- descent haplotypes, called runs of homozygosity (ROH), or estimating fixation index (FST) of genomic windows that indicates genetic differentiation. This study aimed to identify selection signatures in a paternal broiler TT line at generations 7th and 16th of selection and to investigate the genes annotated in these regions as well as the biological pathways involved. For such purpose, ROH and FST-based analysis were performed using whole genome sequence of twenty-eight chickens from two different generations. Results: ROH analysis identified homozygous regions of short and moderate size. Analysis of ROH patterns revealed regions commonly shared among animals and changes in ROH abundance and size between the two generations. Results also suggest that whole genome sequencing (WGS) outperforms SNPchip data avoiding overestimation of ROH size and underestimation of ROH number; however, sequencing costs can limited the number of animals analyzed. FST- based analysis revealed genetic differentiation in several genomic windows. -

Relevance of Translation Initiation in Diffuse Glioma Biology and Its
cells Review Relevance of Translation Initiation in Diffuse Glioma Biology and its Therapeutic Potential Digregorio Marina 1, Lombard Arnaud 1,2, Lumapat Paul Noel 1, Scholtes Felix 1,2, Rogister Bernard 1,3 and Coppieters Natacha 1,* 1 Laboratory of Nervous System Disorders and Therapy, GIGA-Neurosciences Research Centre, University of Liège, 4000 Liège, Belgium; [email protected] (D.M.); [email protected] (L.A.); [email protected] (L.P.N.); [email protected] (S.F.); [email protected] (R.B.) 2 Department of Neurosurgery, CHU of Liège, 4000 Liège, Belgium 3 Department of Neurology, CHU of Liège, 4000 Liège, Belgium * Correspondence: [email protected] Received: 18 October 2019; Accepted: 26 November 2019; Published: 29 November 2019 Abstract: Cancer cells are continually exposed to environmental stressors forcing them to adapt their protein production to survive. The translational machinery can be recruited by malignant cells to synthesize proteins required to promote their survival, even in times of high physiological and pathological stress. This phenomenon has been described in several cancers including in gliomas. Abnormal regulation of translation has encouraged the development of new therapeutics targeting the protein synthesis pathway. This approach could be meaningful for glioma given the fact that the median survival following diagnosis of the highest grade of glioma remains short despite current therapy. The identification of new targets for the development of novel therapeutics is therefore needed in order to improve this devastating overall survival rate. This review discusses current literature on translation in gliomas with a focus on the initiation step covering both the cap-dependent and cap-independent modes of initiation.