Prediction of Biopharmaceutical Drug Disposition Classification System (BDDCS) by Structural Parameters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
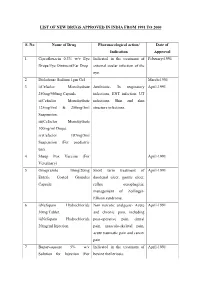
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

An Approach to the Patient with a Dry Mouth
MedicineToday 2014; 15(4): 30-37 PEER REVIEWED FEATURE 2 CPD POINTS An approach to the patient with a dry mouth Key points • The subjective complaint of ELHAM AFLAKI MD; TAHEREH ERFANI MD; NICHOLAS MANOLIOS MB BS(Hons), PhD, MD, FRACP, FRCPA; xerostomia needs to be MARK SCHIFTER FFD, RCSI(Oral Med), FRACDS(Oral Med) differentiated from true salivary hypofunction. Dry mouth is a common and disabling problem. After exclusion of treatable • Salivary hypofunction can significantly reduce quality causes, treatment is symptomatic to prevent the consequences of salivary of life through its adverse hypofunction, such as tooth decay and infection of the oral mucosa. effects on taste, mastication, swallowing, cleansing of the erostomia, or the subjective feeling of neuropathic-induced orofacial dysaesthesia) mouth, killing of microbes a dry mouth, is a common complaint. and psychological and psychiatric disorders, and speech. It is often a consequence of salivary such as anxiety and depression. • Salivary hypofunction is a hypofunction (hyposalivation), in substantive risk factor for X which there is objective evidence of reduced NORMAL SALIVA PRODUCTION dental caries, oral mucosal salivary output or qualitative changes in saliva. Under normal physiological conditions, the disease and infection, Typically, patients complain of oral dryness salivary glands produce 1000 to 1500 mL of particularly oral candidiasis. only when salivary secretion is reduced by more saliva daily as an ultrafiltrate from the circu- • Patients should be than half.1 As saliva has a crucial role in taste lating plasma. Therefore, simple dehydration investigated for contributory perception, mastication, swallowing, cleansing reduces saliva production. The parotid glands and underlying causes, of the mouth, killing of microbes and speech, are the major source of serous saliva (60 to 65% which include drugs and abnormalities in saliva production can signif- of total saliva volume), producing the stimu- rheumatological diseases. -

Metabolic-Hydroxy and Carboxy Functionalization of Alkyl Moieties in Drug Molecules: Prediction of Structure Influence and Pharmacologic Activity
molecules Review Metabolic-Hydroxy and Carboxy Functionalization of Alkyl Moieties in Drug Molecules: Prediction of Structure Influence and Pharmacologic Activity Babiker M. El-Haj 1,* and Samrein B.M. Ahmed 2 1 Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, University of Science and Technology of Fujairah, Fufairah 00971, UAE 2 College of Medicine, Sharjah Institute for Medical Research, University of Sharjah, Sharjah 00971, UAE; [email protected] * Correspondence: [email protected] Received: 6 February 2020; Accepted: 7 April 2020; Published: 22 April 2020 Abstract: Alkyl moieties—open chain or cyclic, linear, or branched—are common in drug molecules. The hydrophobicity of alkyl moieties in drug molecules is modified by metabolic hydroxy functionalization via free-radical intermediates to give primary, secondary, or tertiary alcohols depending on the class of the substrate carbon. The hydroxymethyl groups resulting from the functionalization of methyl groups are mostly oxidized further to carboxyl groups to give carboxy metabolites. As observed from the surveyed cases in this review, hydroxy functionalization leads to loss, attenuation, or retention of pharmacologic activity with respect to the parent drug. On the other hand, carboxy functionalization leads to a loss of activity with the exception of only a few cases in which activity is retained. The exceptions are those groups in which the carboxy functionalization occurs at a position distant from a well-defined primary pharmacophore. Some hydroxy metabolites, which are equiactive with their parent drugs, have been developed into ester prodrugs while carboxy metabolites, which are equiactive to their parent drugs, have been developed into drugs as per se. -

WITHOUTUS010307409B2 (12 ) United States Patent ( 10 ) Patent No
WITHOUTUS010307409B2 (12 ) United States Patent ( 10 ) Patent No. : US 10 , 307 ,409 B2 Chase et al. (45 ) Date of Patent: Jun . 4 , 2019 ( 54 ) MUSCARINIC COMBINATIONS AND THEIR (52 ) U . S . CI. USE FOR COMBATING CPC . .. .. A61K 31/ 4439 (2013 . 01 ) ; A61K 9 /0056 HYPOCHOLINERGIC DISORDERS OF THE (2013 . 01 ) ; A61K 9 / 7023 ( 2013 . 01 ) ; A61K CENTRAL NERVOUS SYSTEM 31 / 166 ( 2013 . 01 ) ; A61K 31 / 216 ( 2013 . 01 ) ; A61K 31 /4178 ( 2013 .01 ) ; A61K 31/ 439 (71 ) Applicant: Chase Pharmaceuticals Corporation , ( 2013 .01 ) ; A61K 31 /44 (2013 . 01 ) ; A61K Washington , DC (US ) 31/ 454 (2013 .01 ) ; A61K 31/ 4725 ( 2013 .01 ) ; A61K 31 /517 (2013 .01 ) ; A61K 45 / 06 ( 72 ) Inventors : Thomas N . Chase , Washington , DC (2013 . 01 ) (US ) ; Kathleen E . Clarence -Smith , ( 58 ) Field of Classification Search Washington , DC (US ) CPC .. A61K 31/ 167 ; A61K 31/ 216 ; A61K 31/ 439 ; A61K 31 /454 ; A61K 31 /4439 ; A61K (73 ) Assignee : Chase Pharmaceuticals Corporation , 31 /4175 ; A61K 31 /4725 Washington , DC (US ) See application file for complete search history. ( * ) Notice : Subject to any disclaimer, the term of this (56 ) References Cited patent is extended or adjusted under 35 U . S . C . 154 (b ) by 0 days . U . S . PATENT DOCUMENTS 5 ,534 ,520 A 7 / 1996 Fisher et al. ( 21) Appl . No. : 15 /260 , 996 2008 /0306103 Al 12 /2008 Fisher et al. 2011/ 0021503 A1* 1/ 2011 Chase . .. A61K 31/ 27 ( 22 ) Filed : Sep . 9 , 2016 514 / 215 2011/ 0071135 A1 * 3 / 2011 Chase . .. .. .. A61K 31/ 166 (65 ) Prior Publication Data 514 / 215 2011 /0245294 Al 10 / 2011 Paborji et al. -

Molecular Mechanisms Underlying Ketamine-Mediated Inhibition Of
Anesthesiology 2005; 102:93–101 © 2004 American Society of Anesthesiologists, Inc. Lippincott Williams & Wilkins, Inc. Molecular Mechanisms Underlying Ketamine-mediated Inhibition of Sarcolemmal Adenosine Triphosphate- sensitive Potassium Channels Takashi Kawano, M.D.,* Shuzo Oshita, M.D.,† Akira Takahashi, M.D.,‡ Yasuo Tsutsumi, M.D.,* Katsuya Tanaka, M.D.,§ Yoshinobu Tomiyama, M.D., Hiroshi Kitahata, M.D.,# Yutaka Nakaya, M.D.** Background: Ketamine inhibits adenosine triphosphate-sen- sensitive channel pore.3 These channels, as metabolic sitive potassium (KATP) channels, which results in the blocking sensors, are associated with such cellular functions as of ischemic preconditioning in the heart and inhibition of va- insulin secretion, cardiac preconditioning, vasodilata- sorelaxation induced by KATP channel openers. In the current 4–7 Downloaded from http://pubs.asahq.org/anesthesiology/article-pdf/102/1/93/357819/0000542-200501000-00017.pdf by guest on 27 September 2021 study, the authors investigated the molecular mechanisms of tion, and neuroprotection. ketamine’s actions on sarcolemmal KATP channels that are re- In cardiac myocytes, intravenous general anesthetics, associated by expressed subunits, inwardly rectifying potas- such as ketamine racemate, propofol, and thiamylal, di- sium channels (Kir6.1 or Kir6.2) and sulfonylurea receptors 8–10 rectly inhibit native sarcolemmal KATP channels. Al- (SUR1, SUR2A, or SUR2B). though these observations suggest that intravenous an- Methods: The authors used inside-out patch clamp configura- tions to investigate the effects of ketamine on the activities of esthetics may impair the endogenous organ protective reassociated Kir6.0/SUR channels containing wild-type, mutant, mechanisms mediated by KATP channels, the possibility or chimeric SURs expressed in COS-7 cells. -

(Trulicity) Pen and the Semaglutide (Ozempic) Pen
Protocol H9X-MC-B021(b) Crossover Study Comparing Dulaglutide (Trulicity) Pen and the Semaglutide (Ozempic) Pen NCT03724981 Approval Date: 30-Nov-2018 H9X-MC-B021(b) Clinical Protocol Page 1 Protocol H9X-MC-B021(b): Crossover Study Comparing the Dulaglutide (Trulicity) Pen and the Semaglutide (Ozempic) Pen Confidential Information The information contained in this document is confidential and is intended for the use of clinical investigators. It is the property of Eli Lilly and Company or its subsidiaries and should not be copied by or distributed to persons not involved in the clinical investigation of dulaglutide (LY2189265), unless such persons are bound by a confidentiality agreement with Eli Lilly and Company or its subsidiaries. Note to Regulatory Authorities: This document may contain protected personal data and/or commercially confidential information exempt from public disclosure. Eli Lilly and Company requests consultation regarding release/redaction prior to any public release. In the United States, this document is subject to Freedom of Information Act (FOIA) Exemption 4 and may not be reproduced or otherwise disseminated without the written approval of Eli Lilly and Company or its subsidiaries. Dulaglutide (LY2189265) Eli Lilly and Company Indianapolis, Indiana USA 46285 Protocol Electronically Signed and Approved by Lilly on 07 Aug 2018 Amendment (a) Electronically Signed and Approved by Lilly on 11 Sep 2018 Amendment (b) Electronically Signed and Approved by Lilly on date below Approval Date: 30-Nov-2018 GMT LY2189265 H9X-MC-B021(b) Clinical Protocol Page 2 Table of Contents Section Page Protocol H9X-MC-B021(b): Crossover Study Comparing the Dulaglutide (Trulicity) Pen and the Semaglutide (Ozempic) Pen ............................................................1 Table of Contents........................................................................................................................2 1. -

Dilantin (Phenytoin Sodium) Extended Oral Capsule Three Times Daily and the Dosage Then Adjusted to Suit Individual Requirements
Dilantin® (Phenytoin Sodium) 100 mg Extended Oral Capsule DESCRIPTION Phenytoin sodium is an antiepileptic drug. Phenytoin sodium is related to the barbiturates in chemical structure, but has a five-membered ring. The chemical name is sodium 5,5-diphenyl-2, 4-imidazolidinedione, having the following structural formula: Each Dilantin— 100 mg Extended Oral Capsule—contains 100 mg phenytoin sodium. Also contains lactose monohydrate, NF; confectioner’s sugar, NF; talc, USP; and magnesium stearate, NF. The capsule body contains titanium dioxide, USP and gelatin, NF. The capsule cap contains FD&C red No. 28; FD&C yellow No. 6; and gelatin NF. Product in vivo performance is characterized by a slow and extended rate of absorption with peak blood concentrations expected in 4 to 12 hours as contrasted to Prompt Phenytoin Sodium Capsules, USP with a rapid rate of absorption with peak blood concentration expected in 1½ to 3 hours. CLINICAL PHARMACOLOGY Phenytoin is an antiepileptic drug which can be used in the treatment of epilepsy. The primary site of action appears to be the motor cortex where spread of seizure activity is inhibited. Possibly by promoting sodium efflux from neurons, phenytoin tends to stabilize the threshold against hyperexcitability caused by excessive stimulation or environmental changes capable of reducing membrane sodium gradient. This includes the reduction of posttetanic potentiation at synapses. Loss of posttetanic potentiation prevents cortical seizure foci from detonating adjacent cortical areas. Phenytoin reduces the maximal activity of brain stem centers responsible for the tonic phase of tonic-clonic (grand mal) seizures. The plasma half-life in man after oral administration of phenytoin averages 22 hours, with a range of 7 to 42 hours. -

2D6 Substrates 2D6 Inhibitors 2D6 Inducers
Physician Guidelines: Drugs Metabolized by Cytochrome P450’s 1 2D6 Substrates Acetaminophen Captopril Dextroamphetamine Fluphenazine Methoxyphenamine Paroxetine Tacrine Ajmaline Carteolol Dextromethorphan Fluvoxamine Metoclopramide Perhexiline Tamoxifen Alprenolol Carvedilol Diazinon Galantamine Metoprolol Perphenazine Tamsulosin Amiflamine Cevimeline Dihydrocodeine Guanoxan Mexiletine Phenacetin Thioridazine Amitriptyline Chloropromazine Diltiazem Haloperidol Mianserin Phenformin Timolol Amphetamine Chlorpheniramine Diprafenone Hydrocodone Minaprine Procainamide Tolterodine Amprenavir Chlorpyrifos Dolasetron Ibogaine Mirtazapine Promethazine Tradodone Aprindine Cinnarizine Donepezil Iloperidone Nefazodone Propafenone Tramadol Aripiprazole Citalopram Doxepin Imipramine Nifedipine Propranolol Trimipramine Atomoxetine Clomipramine Encainide Indoramin Nisoldipine Quanoxan Tropisetron Benztropine Clozapine Ethylmorphine Lidocaine Norcodeine Quetiapine Venlafaxine Bisoprolol Codeine Ezlopitant Loratidine Nortriptyline Ranitidine Verapamil Brofaramine Debrisoquine Flecainide Maprotline olanzapine Remoxipride Zotepine Bufuralol Delavirdine Flunarizine Mequitazine Ondansetron Risperidone Zuclopenthixol Bunitrolol Desipramine Fluoxetine Methadone Oxycodone Sertraline Butylamphetamine Dexfenfluramine Fluperlapine Methamphetamine Parathion Sparteine 2D6 Inhibitors Ajmaline Chlorpromazine Diphenhydramine Indinavir Mibefradil Pimozide Terfenadine Amiodarone Cimetidine Doxorubicin Lasoprazole Moclobemide Quinidine Thioridazine Amitriptyline Cisapride -

The In¯Uence of Medication on Erectile Function
International Journal of Impotence Research (1997) 9, 17±26 ß 1997 Stockton Press All rights reserved 0955-9930/97 $12.00 The in¯uence of medication on erectile function W Meinhardt1, RF Kropman2, P Vermeij3, AAB Lycklama aÁ Nijeholt4 and J Zwartendijk4 1Department of Urology, Netherlands Cancer Institute/Antoni van Leeuwenhoek Hospital, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands; 2Department of Urology, Leyenburg Hospital, Leyweg 275, 2545 CH The Hague, The Netherlands; 3Pharmacy; and 4Department of Urology, Leiden University Hospital, P.O. Box 9600, 2300 RC Leiden, The Netherlands Keywords: impotence; side-effect; antipsychotic; antihypertensive; physiology; erectile function Introduction stopped their antihypertensive treatment over a ®ve year period, because of side-effects on sexual function.5 In the drug registration procedures sexual Several physiological mechanisms are involved in function is not a major issue. This means that erectile function. A negative in¯uence of prescrip- knowledge of the problem is mainly dependent on tion-drugs on these mechanisms will not always case reports and the lists from side effect registries.6±8 come to the attention of the clinician, whereas a Another way of looking at the problem is drug causing priapism will rarely escape the atten- combining available data on mechanisms of action tion. of drugs with the knowledge of the physiological When erectile function is in¯uenced in a negative mechanisms involved in erectile function. The way compensation may occur. For example, age- advantage of this approach is that remedies may related penile sensory disorders may be compen- evolve from it. sated for by extra stimulation.1 Diminished in¯ux of In this paper we will discuss the subject in the blood will lead to a slower onset of the erection, but following order: may be accepted. -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -

List of Union Reference Dates A
Active substance name (INN) EU DLP BfArM / BAH DLP yearly PSUR 6-month-PSUR yearly PSUR bis DLP (List of Union PSUR Submission Reference Dates and Frequency (List of Union Frequency of Reference Dates and submission of Periodic Frequency of submission of Safety Update Reports, Periodic Safety Update 30 Nov. 2012) Reports, 30 Nov. -

THE SPECIFICITY of DRUG BINDING SITES on HUMAN SERUM ALBUMIN Ingvar Sjòholm
THE SPECIFICITY OF DRUG BINDING SITES ON HUMAN SERUM ALBUMIN Ingvar Sjòholm Today, it is well established that the binding of drugs in serum will strongly influence the pharmacokinetic parameters of a drug, such as its distribution volume and clearance. It is also evident that the binding of the drug—in serum and elsewhere in the tissues—will have an influence on the duration and intensity of the pharmacological effect. Several excellent papers and reviews have dealt with these issues in recent years.1"** It is obvious that albumin, being the most abundant protein species in the extracellular fluids, is the most im- portant drug-binding protein, although other proteins can play a pharmacokinetic role. Thus, e.g., orosomucoid (aj-acid glyco- protein) can bind some basic and neutral drugs ,9 and lipoproteins some highly hydrophobic drugs.10 The primary structure of human serum albumin (HSA) is now known.1:L'^2 However, all efforts to study the three-dimensional structure by x-ray spectroscopy have hitherto failed, and a detailed knowledge of the mechanisms involved in the binding of drugs or endogenous compounds is still missing. The broad binding specificity of HSA is remarkable. Several compounds of widely different struc- ture can be bound with high affinity—e.g., fatty acids, bilirubin, tryptophan, as well as many drugs. It is also striking that different reports from quantitative studies on the binding of different com- pounds have shown varying results, which cannot be solely explained by technical problems or different experimental conditions. All avail- able information indicates that HSA is a highly "flexible" and "adapt- able" molecule, the structure of which can be strongly influenced by different "modulating" substances.