LIONS: Analysis Suite for Detecting and Quantifying Transposable Element Initiated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

The Nuclear Localization Pattern and Interaction Partners of GTF2IRD1 Demonstrate a Role in Chromatin Regulation
Hum Genet DOI 10.1007/s00439-015-1591-0 ORIGINAL INVESTIGATION The nuclear localization pattern and interaction partners of GTF2IRD1 demonstrate a role in chromatin regulation Paulina Carmona‑Mora1 · Jocelyn Widagdo2 · Florence Tomasetig1 · Cesar P. Canales1 · Yeojoon Cha1 · Wei Lee1 · Abdullah Alshawaf3 · Mirella Dottori3 · Renee M. Whan4 · Edna C. Hardeman1 · Stephen J. Palmer1 Received: 11 February 2015 / Accepted: 4 August 2015 © Springer-Verlag Berlin Heidelberg 2015 Abstract GTF2IRD1 is one of the three members of the mostly involved in chromatin modification and transcrip- GTF2I gene family, clustered on chromosome 7 within a tional regulation, whilst others indicate an unexpected role 1.8 Mb region that is prone to duplications and deletions in connection with the primary cilium. Mapping of the sites in humans. Hemizygous deletions cause Williams–Beuren of protein interaction also indicates key features regarding syndrome (WBS) and duplications cause WBS duplica- the evolution of the GTF2IRD1 protein. These data provide tion syndrome. These copy number variations disturb a a visual and molecular basis for GTF2IRD1 nuclear func- variety of developmental systems and neurological func- tion that will lead to an understanding of its role in brain, tions. Human mapping data and analyses of knockout mice behaviour and human disease. show that GTF2IRD1 and GTF2I underpin the craniofacial abnormalities, mental retardation, visuospatial deficits and Abbreviations hypersociability of WBS. However, the cellular role of the hESC Human embryonic stem cells GTF2IRD1 protein is poorly understood due to its very PLA Proximity ligation assay low abundance and a paucity of reagents. Here, for the first STED Stimulated emission depletion time, we show that endogenous GTF2IRD1 has a punctate WBS Williams–Beuren syndrome pattern in the nuclei of cultured human cell lines and neu- Y2H Yeast two-hybrid rons. -

Placenta-Derived Exosomes Continuously Increase in Maternal
Sarker et al. Journal of Translational Medicine 2014, 12:204 http://www.translational-medicine.com/content/12/1/204 RESEARCH Open Access Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy Suchismita Sarker1, Katherin Scholz-Romero1, Alejandra Perez2, Sebastian E Illanes1,2,3, Murray D Mitchell1, Gregory E Rice1,2 and Carlos Salomon1,2* Abstract Background: Human placenta releases specific nanovesicles (i.e. exosomes) into the maternal circulation during pregnancy, however, the presence of placenta-derived exosomes in maternal blood during early pregnancy remains to be established. The aim of this study was to characterise gestational age related changes in the concentration of placenta-derived exosomes during the first trimester of pregnancy (i.e. from 6 to 12 weeks) in plasma from women with normal pregnancies. Methods: A time-series experimental design was used to establish pregnancy-associated changes in maternal plasma exosome concentrations during the first trimester. A series of plasma were collected from normal healthy women (10 patients) at 6, 7, 8, 9, 10, 11 and 12 weeks of gestation (n = 70). We measured the stability of these vesicles by quantifying and observing their protein and miRNA contents after the freeze/thawing processes. Exosomes were isolated by differential and buoyant density centrifugation using a sucrose continuous gradient and characterised by their size distribution and morphology using the nanoparticles tracking analysis (NTA; Nanosight™) and electron microscopy (EM), respectively. The total number of exosomes and placenta-derived exosomes were determined by quantifying the immunoreactive exosomal marker, CD63 and a placenta-specific marker (Placental Alkaline Phosphatase PLAP). -

Curcumin Alters Gene Expression-Associated DNA Damage, Cell Cycle, Cell Survival and Cell Migration and Invasion in NCI-H460 Human Lung Cancer Cells in Vitro
ONCOLOGY REPORTS 34: 1853-1874, 2015 Curcumin alters gene expression-associated DNA damage, cell cycle, cell survival and cell migration and invasion in NCI-H460 human lung cancer cells in vitro I-TSANG CHIANG1,2, WEI-SHU WANG3, HSIN-CHUNG LIU4, SU-TSO YANG5, NOU-YING TANG6 and JING-GUNG CHUNG4,7 1Department of Radiation Oncology, National Yang‑Ming University Hospital, Yilan 260; 2Department of Radiological Technology, Central Taiwan University of Science and Technology, Taichung 40601; 3Department of Internal Medicine, National Yang‑Ming University Hospital, Yilan 260; 4Department of Biological Science and Technology, China Medical University, Taichung 404; 5Department of Radiology, China Medical University Hospital, Taichung 404; 6Graduate Institute of Chinese Medicine, China Medical University, Taichung 404; 7Department of Biotechnology, Asia University, Taichung 404, Taiwan, R.O.C. Received March 31, 2015; Accepted June 26, 2015 DOI: 10.3892/or.2015.4159 Abstract. Lung cancer is the most common cause of cancer CARD6, ID1 and ID2 genes, associated with cell survival and mortality and new cases are on the increase worldwide. the BRMS1L, associated with cell migration and invasion. However, the treatment of lung cancer remains unsatisfactory. Additionally, 59 downregulated genes exhibited a >4-fold Curcumin has been shown to induce cell death in many human change, including the DDIT3 gene, associated with DNA cancer cells, including human lung cancer cells. However, the damage; while 97 genes had a >3- to 4-fold change including the effects of curcumin on genetic mechanisms associated with DDIT4 gene, associated with DNA damage; the CCPG1 gene, these actions remain unclear. Curcumin (2 µM) was added associated with cell cycle and 321 genes with a >2- to 3-fold to NCI-H460 human lung cancer cells and the cells were including the GADD45A and CGREF1 genes, associated with incubated for 24 h. -

Looking for Missing Proteins in the Proteome Of
Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update Yves Vandenbrouck, Lydie Lane, Christine Carapito, Paula Duek, Karine Rondel, Christophe Bruley, Charlotte Macron, Anne Gonzalez de Peredo, Yohann Coute, Karima Chaoui, et al. To cite this version: Yves Vandenbrouck, Lydie Lane, Christine Carapito, Paula Duek, Karine Rondel, et al.. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. Journal of Proteome Research, American Chemical Society, 2016, 15 (11), pp.3998-4019. 10.1021/acs.jproteome.6b00400. hal-02191502 HAL Id: hal-02191502 https://hal.archives-ouvertes.fr/hal-02191502 Submitted on 19 Mar 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Journal of Proteome Research 1 2 3 Looking for missing proteins in the proteome of human spermatozoa: an 4 update 5 6 Yves Vandenbrouck1,2,3,#,§, Lydie Lane4,5,#, Christine Carapito6, Paula Duek5, Karine Rondel7, 7 Christophe Bruley1,2,3, Charlotte Macron6, Anne Gonzalez de Peredo8, Yohann Couté1,2,3, 8 Karima Chaoui8, Emmanuelle Com7, Alain Gateau5, AnneMarie Hesse1,2,3, Marlene 9 Marcellin8, Loren Méar7, Emmanuelle MoutonBarbosa8, Thibault Robin9, Odile Burlet- 10 Schiltz8, Sarah Cianferani6, Myriam Ferro1,2,3, Thomas Fréour10,11, Cecilia Lindskog12,Jérôme 11 1,2,3 7,§ 12 Garin , Charles Pineau . -

Supplemental Table S1. Primers for Sybrgreen Quantitative RT-PCR Assays
Supplemental Table S1. Primers for SYBRGreen quantitative RT-PCR assays. Gene Accession Primer Sequence Length Start Stop Tm GC% GAPDH NM_002046.3 GAPDH F TCCTGTTCGACAGTCAGCCGCA 22 39 60 60.43 59.09 GAPDH R GCGCCCAATACGACCAAATCCGT 23 150 128 60.12 56.52 Exon junction 131/132 (reverse primer) on template NM_002046.3 DNAH6 NM_001370.1 DNAH6 F GGGCCTGGTGCTGCTTTGATGA 22 4690 4711 59.66 59.09% DNAH6 R TAGAGAGCTTTGCCGCTTTGGCG 23 4797 4775 60.06 56.52% Exon junction 4790/4791 (reverse primer) on template NM_001370.1 DNAH7 NM_018897.2 DNAH7 F TGCTGCATGAGCGGGCGATTA 21 9973 9993 59.25 57.14% DNAH7 R AGGAAGCCATGTACAAAGGTTGGCA 25 10073 10049 58.85 48.00% Exon junction 9989/9990 (forward primer) on template NM_018897.2 DNAI1 NM_012144.2 DNAI1 F AACAGATGTGCCTGCAGCTGGG 22 673 694 59.67 59.09 DNAI1 R TCTCGATCCCGGACAGGGTTGT 22 822 801 59.07 59.09 Exon junction 814/815 (reverse primer) on template NM_012144.2 RPGRIP1L NM_015272.2 RPGRIP1L F TCCCAAGGTTTCACAAGAAGGCAGT 25 3118 3142 58.5 48.00% RPGRIP1L R TGCCAAGCTTTGTTCTGCAAGCTGA 25 3238 3214 60.06 48.00% Exon junction 3124/3125 (forward primer) on template NM_015272.2 Supplemental Table S2. Transcripts that differentiate IPF/UIP from controls at 5%FDR Fold- p-value Change Transcript Gene p-value p-value p-value (IPF/UIP (IPF/UIP Cluster ID RefSeq Symbol gene_assignment (Age) (Gender) (Smoking) vs. C) vs. C) NM_001178008 // CBS // cystathionine-beta- 8070632 NM_001178008 CBS synthase // 21q22.3 // 875 /// NM_0000 0.456642 0.314761 0.418564 4.83E-36 -2.23 NM_003013 // SFRP2 // secreted frizzled- 8103254 NM_003013 -

Serum APOA4 Pharmacodynamically Represents Administered Recombinant Human Hepatocyte Growth Factor (E3112)
International Journal of Molecular Sciences Article Serum APOA4 Pharmacodynamically Represents Administered Recombinant Human Hepatocyte Growth Factor (E3112) Sotaro Motoi 1,2, Mai Uesugi 3, Takashi Obara 3, Katsuhiro Moriya 2, Yoshihisa Arita 2, Hideaki Ogasawara 2, Motohiro Soejima 1,2, Toshio Imai 2 and Tetsu Kawano 2,* 1 Eisai Product Creation Systems, KAN Product Creation Unit, Eisai Co., Ltd., 5-1-3 Tokodai, Tsukuba, Ibaraki 3002635, Japan; [email protected] (S.M.); [email protected] (M.S.) 2 KAN Research Institute, Inc., 6-8-2 Minatojima-Minamimachi, Chuo-Ku, Kobe, Hyogo 6500047, Japan; [email protected] (K.M.); [email protected] (Y.A.); [email protected] (H.O.); [email protected] (T.I.) 3 Medicine Creation, Neurology Business Group, Translational Medicine Department, Eisai Co., Ltd., 5-1-3 Tokodai, Tsukuba, Ibaraki 3002635, Japan; [email protected] (M.U.); [email protected] (T.O.) * Correspondence: [email protected]; Tel.: +81-78-306-5910; Fax: +81-78-306-5920 Abstract: Background: Hepatocyte growth factor (HGF) is an endogenously induced bioactive molecule that has strong anti-apoptotic and tissue repair activities. In this research, we identified APOA4 as a novel pharmacodynamic (PD) marker of the recombinant human HGF (rh-HGF), E3112. Methods: rh-HGF was administered to mice, and their livers were investigated for the PD marker. Candidates were identified from soluble proteins and validated by using human hepatocytes in vitro and an animal disease model in vivo, in which its c-Met dependency was also ensured. -
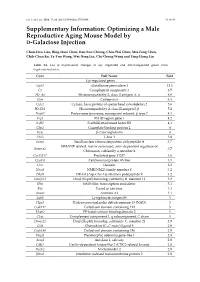
Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection
Int. J. Mol. Sci. 2016, 17, 98; doi:10.3390/ijms17010098 S1 of S4 Supplementary Information: Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection Chun-Hou Liao, Bing-Huei Chen, Han-Sun Chiang, Chiu-Wei Chen, Mei-Feng Chen, Chih-Chun Ke, Ya-Yun Wang, Wei-Ning Lin, Chi-Chung Wang and Ying-Hung Lin Table S1. List of expressional changes of up- regulated and down-regulated genes from D-gal-injected mice. Gene Full Name Fold Up-regulated genes Gpx3 Glutathione peroxidase 3 13.1 C3 Complement component 3 8.5 H2-Aa Histocompatibility 2, class II antigen A, α 6.9 Ctss Cathepsin S 6.3 Cylc2 Cylicin, basic protein of sperm head cytoskeleton 2 5.6 H2-Eb1 Histocompatibility 2, class II antigen E β 5.2 Psmb7 Proteasome (prosome, macropain) subunit, β type 7 4.3 Frg1 FSHD region gene 1 4.2 Safb2 Scaffold attachment factor B2 4.1 Gbp2 Guanylate binding protein 2 4 B2m β-2 microglobulin 4 Tbx3 T-box 3 3.8 Snrpa Small nuclear ribonucleoprotein polypeptide A 3.7 SWI/SNF related, matrix associated, actin dependent regulator of Smarca2 3.7 Chromatin, subfamily a, member 2 Gm13237 Predicted gene 13237 3.6 Cep85l Centrosomal protein 85-like 3.5 Dcn Decorin 3.2 Nme8 NME/NM23 family member 8 3.2 Dhx9 DEAH (Asp-Glu-Ala-His) box polypeptide 9 3.2 Dnajb11 DnaJ (Hsp40) homolog, subfamily B, member 11 3.2 Sltm SAFB-like, transcription modulator 3.1 Fus Fused in sarcoma 3.1 Anxa1 Annexin A1 3.1 Ly86 Lymphocyte antigen 86 3 Hpgd Hydroxyprostaglandin dehydrogenase 15 (NAD) 3 Ccdc112 Coiled-coil domain containing 112 3 Efcab2 EF-hand calcium binding domain 2 3 C1qc Complement component 1, q subcomponent, C chain 3 Dnajc21 DnaJ (Hsp40) homolog, subfamily C, member 21 2.9 Ccl8 Chemokine (C–C motif) ligand 8 2.9 Ccdc186 Coiled-coil domain containing 186 2.9 Plagl1 Pleomorphic adenoma gene-like 1 2.8 Amy1 Amylase 1, salivary 2.8 Cdkl2 Cyclin-dependent kinase-like 2 (CDC2-related kinase) 2.8 Nucb2 Nucleobindin 2 2.8 Ifitm2 Interferon induced transmembrane protein 2 2.8 Int. -

Comprehensive Analysis Reveals Novel Gene Signature in Head and Neck Squamous Cell Carcinoma: Predicting Is Associated with Poor Prognosis in Patients
5892 Original Article Comprehensive analysis reveals novel gene signature in head and neck squamous cell carcinoma: predicting is associated with poor prognosis in patients Yixin Sun1,2#, Quan Zhang1,2#, Lanlin Yao2#, Shuai Wang3, Zhiming Zhang1,2 1Department of Breast Surgery, The First Affiliated Hospital of Xiamen University, School of Medicine, Xiamen University, Xiamen, China; 2School of Medicine, Xiamen University, Xiamen, China; 3State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, China Contributions: (I) Conception and design: Y Sun, Q Zhang; (II) Administrative support: Z Zhang; (III) Provision of study materials or patients: Y Sun, Q Zhang; (IV) Collection and assembly of data: Y Sun, L Yao; (V) Data analysis and interpretation: Y Sun, S Wang; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. #These authors contributed equally to this work. Correspondence to: Zhiming Zhang. Department of Surgery, The First Affiliated Hospital of Xiamen University, Xiamen, China. Email: [email protected]. Background: Head and neck squamous cell carcinoma (HNSC) remains an important public health problem, with classic risk factors being smoking and excessive alcohol consumption and usually has a poor prognosis. Therefore, it is important to explore the underlying mechanisms of tumorigenesis and screen the genes and pathways identified from such studies and their role in pathogenesis. The purpose of this study was to identify genes or signal pathways associated with the development of HNSC. Methods: In this study, we downloaded gene expression profiles of GSE53819 from the Gene Expression Omnibus (GEO) database, including 18 HNSC tissues and 18 normal tissues. -

Investigating the Role of the O-Glcnac Modification During the Neural Differentiation of Human Embryonic Stem Cells
Investigating the role of the O-GlcNAc modification during the neural differentiation of human embryonic stem cells By Lissette Andres A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Molecular and Cell Biology in the Graduate Division of the University of California, Berkeley Committee in charge: Professor Carolyn R. Bertozzi, Chair Professor Michael Rape Professor Ellen Robey Professor David V. Schaffer Fall 2014 Investigating the role of the O-GlcNAc modification during the neural differentiation of human embryonic stem cells © 2014 by Lissette Andres ! ! Abstract Investigating the role of the O-GlcNAc modification during the neural differentiation of human embryonic stem cells by Lissette Andres Doctor of Philosophy in Molecular and Cell Biology University of California, Berkeley Professor Carolyn R. Bertozzi, Chair Human embryonic stem cells (hESCs) have the ability of propagating indefinitely and can be induced to differentiate into specialized cell types; however, the molecular mechanisms that govern the self-renewal and conversion of hESCs into a variety of cell types are not well understood. In order to understand the molecular mechanisms involved in the modulation of these processes it is necessary to look beyond what is encoded in the genome, and look into other forms of cellular regulation such as post- translational modifications. The addition of a single monosaccharide, !-N- acetylglucosamine (O-GlcNAc), to the hydroxy side chain of serine or threonine residues of protein is an intracellular, post-translational modification that shares qualities with phosphorylation. The enzymes involved in the addition and removal of O-GlcNAc, O-GlcNAc transferase and O-GlcNAcase, respectively, have been shown to target key transcriptional and epigenetic regulators. -

The Role of Lamin Associated Domains in Global Chromatin Organization and Nuclear Architecture
THE ROLE OF LAMIN ASSOCIATED DOMAINS IN GLOBAL CHROMATIN ORGANIZATION AND NUCLEAR ARCHITECTURE By Teresa Romeo Luperchio A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland March 2016 © 2016 Teresa Romeo Luperchio All Rights Reserved ABSTRACT Nuclear structure and scaffolding have been implicated in expression and regulation of the genome (Elcock and Bridger 2010; Fedorova and Zink 2008; Ferrai et al. 2010; Li and Reinberg 2011; Austin and Bellini 2010). Discrete domains of chromatin exist within the nuclear volume, and are suggested to be organized by patterns of gene activity (Zhao, Bodnar, and Spector 2009). The nuclear periphery, which consists of the inner nuclear membrane and associated proteins, forms a sub- nuclear compartment that is mostly associated with transcriptionally repressed chromatin and low gene expression (Guelen et al. 2008). Previous studies from our lab and others have shown that repositioning genes to the nuclear periphery is sufficient to induce transcriptional repression (K L Reddy et al. 2008; Finlan et al. 2008). In addition, a number of studies have provided evidence that many tissue types, including muscle, brain and blood, use the nuclear periphery as a compartment during development to regulate expression of lineage specific genes (Meister et al. 2010; Szczerbal, Foster, and Bridger 2009; Yao et al. 2011; Kosak et al. 2002; Peric-Hupkes et al. 2010). These large regions of chromatin that come in molecular contact with the nuclear periphery are called Lamin Associated Domains (LADs). The studies described in this dissertation have furthered our understanding of maintenance and establishment of LADs as well as the relationship of LADs with the epigenome and other factors that influence three-dimensional chromatin structure. -

Supplemental Solier
Supplementary Figure 1. Importance of Exon numbers for transcript downregulation by CPT Numbers of down-regulated genes for four groups of comparable size genes, differing only by the number of exons. Supplementary Figure 2. CPT up-regulates the p53 signaling pathway genes A, List of the GO categories for the up-regulated genes in CPT-treated HCT116 cells (p<0.05). In bold: GO category also present for the genes that are up-regulated in CPT- treated MCF7 cells. B, List of the up-regulated genes in both CPT-treated HCT116 cells and CPT-treated MCF7 cells (CPT 4 h). C, RT-PCR showing the effect of CPT on JUN and H2AFJ transcripts. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. Supplementary Figure 3. Down-regulation of RNA degradation-related genes after CPT treatment A, “RNA degradation” pathway from KEGG. The genes with “red stars” were down- regulated genes after CPT treatment. B, Affy Exon array data for the “CNOT” genes. The log2 difference for the “CNOT” genes expression depending on CPT treatment was normalized to the untreated controls. C, RT-PCR showing the effect of CPT on “CNOT” genes down-regulation. HCT116 cells were treated with CPT (10 µM, 20 h) and CNOT6L, CNOT2, CNOT4 and CNOT6 mRNA were analysed by RT-PCR. Control cells were exposed to DMSO. β2 microglobulin (β2) mRNA was used as control. D, CNOT6L down-regulation after CPT treatment. CNOT6L transcript was analysed by Q- PCR. Supplementary Figure 4. Down-regulation of ubiquitin-related genes after CPT treatment A, “Ubiquitin-mediated proteolysis” pathway from KEGG.