Determination of Thermodynamic and Structural Quantities of Polymers by Scattering Techniques
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -
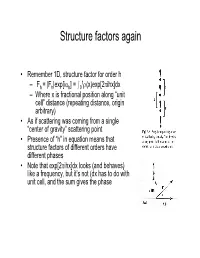
Structure Factors Again
Structure factors again • Remember 1D, structure factor for order h 1 –Fh = |Fh|exp[iαh] = I0 ρ(x)exp[2πihx]dx – Where x is fractional position along “unit cell” distance (repeating distance, origin arbitrary) • As if scattering was coming from a single “center of gravity” scattering point • Presence of “h” in equation means that structure factors of different orders have different phases • Note that exp[2πihx]dx looks (and behaves) like a frequency, but it’s not (dx has to do with unit cell, and the sum gives the phase Back and Forth • Fourier sez – For any function f(x), there is a “transform” of it which is –F(h) = Ûf(x)exp(2pi(hx))dx – Where h is reciprocal of x (1/x) – Structure factors look like that • And it works backward –f(x) = ÛF(h)exp(-2pi(hx))dh – Or, if h comes only in discrete points –f(x) = SF(h)exp(-2pi(hx)) Structure factors, cont'd • Structure factors are a "Fourier transform" - a sum of components • Fourier transforms are reversible – From summing distribution of ρ(x), get hth order of diffraction – From summing hth orders of diffraction, get back ρ(x) = Σ Fh exp[-2πihx] Two dimensional scattering • In Frauenhofer diffraction (1D), we considered scattering from points, along the line • In 2D diffraction, scattering would occur from lines. • Numbering of the lines by where they cut the edges of a unit cell • Atom density in various lines can differ • Reflections now from planes Extension to 3D – Planes defined by extension from 2D case – Unit cells differ • Depends on arrangement of materials in 3D lattice • = "Space -

System Design and Verification of the Precession Electron Diffraction Technique
NORTHWESTERN UNIVERSITY System Design and Verification of the Precession Electron Diffraction Technique A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS for the degree DOCTOR OF PHILOSOPHY Field of Materials Science and Engineering By Christopher Su-Yan Own EVANSTON, ILLINOIS First published on the WWW 01, August 2005 Build 05.12.07. PDF available for download at: http://www.numis.northwestern.edu/Research/Current/precession.shtml c Copyright by Christopher Su-Yan Own 2005 All Rights Reserved ii ABSTRACT System Design and Verification of the Precession Electron Diffraction Technique Christopher Su-Yan Own Bulk structural crystallography is generally a two-part process wherein a rough starting structure model is first derived, then later refined to give an accurate model of the structure. The critical step is the deter- mination of the initial model. As materials problems decrease in length scale, the electron microscope has proven to be a versatile and effective tool for studying many problems. However, study of complex bulk structures by electron diffraction has been hindered by the problem of dynamical diffraction. This phenomenon makes bulk electron diffraction very sensitive to specimen thickness, and expensive equip- ment such as aberration-corrected scanning transmission microscopes or elaborate methodology such as high resolution imaging combined with diffraction and simulation are often required to generate good starting structures. The precession electron diffraction technique (PED), which has the ability to significantly reduce dynamical effects in diffraction patterns, has shown promise as being a “philosopher’s stone” for bulk electron diffraction. However, a comprehensive understanding of its abilities and limitations is necessary before it can be put into widespread use as a standalone technique. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and Inano Center University of Aarhus Denmark SAXS Lab
Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and iNANO Center University of Aarhus Denmark SAXS lab 1 Outline • Model fitting and least-squares methods • Available form factors ex: sphere, ellipsoid, cylinder, spherical subunits… ex: polymer chain • Monte Carlo integration for form factors of complex structures • Monte Carlo simulations for form factors of polymer models • Concentration effects and structure factors Zimm approach Spherical particles Elongated particles (approximations) Polymers 2 Motivation - not to replace shape reconstruction and crystal-structure based modeling – we use the methods extensively - alternative approaches to reduce the number of degrees of freedom in SAS data structural analysis (might make you aware of the limited information content of your data !!!) - provide polymer-theory based modeling of flexible chains - describe and correct for concentration effects 3 Literature Jan Skov Pedersen, Analysis of Small-Angle Scattering Data from Colloids and Polymer Solutions: Modeling and Least-squares Fitting (1997). Adv. Colloid Interface Sci. , 70 , 171-210. Jan Skov Pedersen Monte Carlo Simulation Techniques Applied in the Analysis of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier Science B.V. p. 381 Jan Skov Pedersen Modelling of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier -
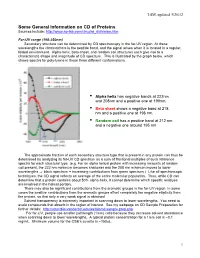
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Direct Phase Determination in Protein Electron Crystallography
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 1791–1794, March 1997 Biophysics Direct phase determination in protein electron crystallography: The pseudo-atom approximation (electron diffractionycrystal structure analysisydirect methodsymembrane proteins) DOUGLAS L. DORSET Electron Diffraction Department, Hauptman–Woodward Medical Research Institute, Inc., 73 High Street, Buffalo, NY 14203-1196 Communicated by Herbert A. Hauptman, Hauptman–Woodward Medical Research Institute, Buffalo, NY, December 12, 1996 (received for review October 28, 1996) ABSTRACT The crystal structure of halorhodopsin is Another approach to such phasing problems, especially in determined directly in its centrosymmetric projection using cases where the structures have appropriate distributions of 6.0-Å-resolution electron diffraction intensities, without in- mass, would be to adopt a pseudo-atom approach. The concept cluding any previous phase information from the Fourier of using globular sub-units as quasi-atoms was discussed by transform of electron micrographs. The potential distribution David Harker in 1953, when he showed that an appropriate in the projection is assumed a priori to be an assembly of globular scattering factor could be used to normalize the globular densities. By an appropriate dimensional re-scaling, low-resolution diffraction intensities with higher accuracy than these ‘‘globs’’ are then assumed to be pseudo-atoms for the actual atomic scattering factors employed for small mol- normalization of the observed structure factors. After this ecule structures -

Protein Folding and Structure Prediction
Protein Folding and Structure Prediction A Statistician's View Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Proteins Amino acids without peptide bonds. Amino acids with peptide bonds. ¡ Amino acids are the building blocks of proteins. Proteins Both figures show the same protein (the bacterial protein L). The right figure also highlights the secondary structure elements. Space Resolution limit of a light microscope Glucose Ribosome Red blood cell C−C bond Hemoglobin Bacterium 1 10 100 1000 10000 100000 1nm 1µm Distance [ A° ] Energy C−C bond Green Noncovalent bond light Glucose Thermal ATP 0.1 1 10 100 1000 Energy [ kcal/mol ] Non-Bonding Interactions Amino acids of a protein are joined by covalent bonding interactions. The polypep- tide is folded in three dimension by non-bonding interactions. These interactions, which can easily be disrupted by extreme pH, temperature, pressure, and denatu- rants, are: Electrostatic Interactions (5 kcal/mol) Hydrogen-bond Interactions (3-7 kcal/mol) Van Der Waals Interactions (1 kcal/mol) ¡ Hydrophobic Interactions ( 10 kcal/mol) The total inter-atomic force acting between two atoms is the sum of all the forces they exert on each other. Energy Profile Transition State Denatured State Native State Radius of Gyration of Denatured Proteins Do chemically denatured proteins behave as random coils? The radius of gyration Rg of a protein is defined as the root mean square dis- tance from each atom of the protein to their centroid. For an ideal (infinitely thin) random-coil chain in a solvent, the average radius 0.5 ¡ of gyration of a random coil is a simple function of its length n: Rg n For an excluded volume polymer (a polymer with non-zero thickness and non- trivial interactions between monomers) in a solvent, the average radius of gyra- 0.588 tion, we have Rg n (Flory 1953). -

Solid State Physics: Problem Set #3 Structural Determination Via X-Ray Scattering Due: Friday Jan
Physics 375 Fall (12) 2003 Solid State Physics: Problem Set #3 Structural Determination via X-Ray Scattering Due: Friday Jan. 31 by 6 pm Reading assignment: for Monday, 3.2-3.3 (structure factors for different lattices) for Wednesday, 3.4-3.7 (scattering methods) for Friday, 4.1-4.3 (mechanical properties of crystals) Problem assignment: Chapter 3 Problems: *3.1 Debye-Scherrer analysis (identify the crystal structure) [Robert] 3.2 Lattice parameter from Debye-Scherrer data 3.11 Measuring thermal expansion with X-ray scattering A1. The unit cell dimension of fcc copper is 0.36 nm. Calculate the longest wavelength of X- rays which will produce diffraction from the close packed planes. From what planes could X- rays with l=0.50 nm be diffracted? *A2. Single crystal diffraction: A cubic crystal with lattice spacing 0.4 nm is mounted with its [001] axis perpendicular to an incident X-ray beam of wavelength 0.1 nm. Initially the crystal is set so as to produce a diffracted beam associated with the (020) planes. Calculate the angle through which the crystal must be turned in order to produce a beam from the (hkl) planes where: a) (hkl)=(030) b) (hkl)=(130) [Brian] c) Which of these diffracted beams would be forbidden if the crystal is: i) sc; ii) bcc; iii) fcc A3. Structure factors for the fcc and diamond bases. a) Construct the structure factor Shkl for the fcc lattice and show that it vanishes unless h, k, and l are all even or all odd. b) Construct the structure factor Shkl for the diamond lattice and show that it vanishes unless h, k, and l are all odd or h+k+l=4n where n is an integer. -
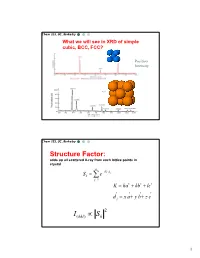
Simple Cubic Lattice
Chem 253, UC, Berkeley What we will see in XRD of simple cubic, BCC, FCC? Position Intensity Chem 253, UC, Berkeley Structure Factor: adds up all scattered X-ray from each lattice points in crystal n iKd j Sk e j1 K ha kb lc d j x a y b z c 2 I(hkl) Sk 1 Chem 253, UC, Berkeley X-ray scattered from each primitive cell interfere constructively when: eiKR 1 2d sin n For n-atom basis: sum up the X-ray scattered from the whole basis Chem 253, UC, Berkeley ' k d k d di R j ' K k k Phase difference: K (di d j ) The amplitude of the two rays differ: eiK(di d j ) 2 Chem 253, UC, Berkeley The amplitude of the rays scattered at d1, d2, d3…. are in the ratios : eiKd j The net ray scattered by the entire cell: n iKd j Sk e j1 2 I(hkl) Sk Chem 253, UC, Berkeley For simple cubic: (0,0,0) iK0 Sk e 1 3 Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis 1 2 iK ( x y z ) iKd j iK0 2 Sk e e e j1 1 ei (hk l) 1 (1)hkl S=2, when h+k+l even S=0, when h+k+l odd, systematical absence Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis S=2, when h+k+l even S=0, when h+k+l odd, systematical absence (100): destructive (200): constructive 4 Chem 253, UC, Berkeley Observable diffraction peaks h2 k 2 l 2 Ratio SC: 1,2,3,4,5,6,8,9,10,11,12.