Structure Factors Again
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

System Design and Verification of the Precession Electron Diffraction Technique
NORTHWESTERN UNIVERSITY System Design and Verification of the Precession Electron Diffraction Technique A DISSERTATION SUBMITTED TO THE GRADUATE SCHOOL IN PARTIAL FULFILLMENT OF THE REQUIREMENTS for the degree DOCTOR OF PHILOSOPHY Field of Materials Science and Engineering By Christopher Su-Yan Own EVANSTON, ILLINOIS First published on the WWW 01, August 2005 Build 05.12.07. PDF available for download at: http://www.numis.northwestern.edu/Research/Current/precession.shtml c Copyright by Christopher Su-Yan Own 2005 All Rights Reserved ii ABSTRACT System Design and Verification of the Precession Electron Diffraction Technique Christopher Su-Yan Own Bulk structural crystallography is generally a two-part process wherein a rough starting structure model is first derived, then later refined to give an accurate model of the structure. The critical step is the deter- mination of the initial model. As materials problems decrease in length scale, the electron microscope has proven to be a versatile and effective tool for studying many problems. However, study of complex bulk structures by electron diffraction has been hindered by the problem of dynamical diffraction. This phenomenon makes bulk electron diffraction very sensitive to specimen thickness, and expensive equip- ment such as aberration-corrected scanning transmission microscopes or elaborate methodology such as high resolution imaging combined with diffraction and simulation are often required to generate good starting structures. The precession electron diffraction technique (PED), which has the ability to significantly reduce dynamical effects in diffraction patterns, has shown promise as being a “philosopher’s stone” for bulk electron diffraction. However, a comprehensive understanding of its abilities and limitations is necessary before it can be put into widespread use as a standalone technique. -

Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and Inano Center University of Aarhus Denmark SAXS Lab
Form and Structure Factors: Modeling and Interactions Jan Skov Pedersen, Department of Chemistry and iNANO Center University of Aarhus Denmark SAXS lab 1 Outline • Model fitting and least-squares methods • Available form factors ex: sphere, ellipsoid, cylinder, spherical subunits… ex: polymer chain • Monte Carlo integration for form factors of complex structures • Monte Carlo simulations for form factors of polymer models • Concentration effects and structure factors Zimm approach Spherical particles Elongated particles (approximations) Polymers 2 Motivation - not to replace shape reconstruction and crystal-structure based modeling – we use the methods extensively - alternative approaches to reduce the number of degrees of freedom in SAS data structural analysis (might make you aware of the limited information content of your data !!!) - provide polymer-theory based modeling of flexible chains - describe and correct for concentration effects 3 Literature Jan Skov Pedersen, Analysis of Small-Angle Scattering Data from Colloids and Polymer Solutions: Modeling and Least-squares Fitting (1997). Adv. Colloid Interface Sci. , 70 , 171-210. Jan Skov Pedersen Monte Carlo Simulation Techniques Applied in the Analysis of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier Science B.V. p. 381 Jan Skov Pedersen Modelling of Small-Angle Scattering Data from Colloids and Polymer Systems in Neutrons, X-Rays and Light P. Lindner and Th. Zemb (Editors) 2002 Elsevier -

Direct Phase Determination in Protein Electron Crystallography
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 1791–1794, March 1997 Biophysics Direct phase determination in protein electron crystallography: The pseudo-atom approximation (electron diffractionycrystal structure analysisydirect methodsymembrane proteins) DOUGLAS L. DORSET Electron Diffraction Department, Hauptman–Woodward Medical Research Institute, Inc., 73 High Street, Buffalo, NY 14203-1196 Communicated by Herbert A. Hauptman, Hauptman–Woodward Medical Research Institute, Buffalo, NY, December 12, 1996 (received for review October 28, 1996) ABSTRACT The crystal structure of halorhodopsin is Another approach to such phasing problems, especially in determined directly in its centrosymmetric projection using cases where the structures have appropriate distributions of 6.0-Å-resolution electron diffraction intensities, without in- mass, would be to adopt a pseudo-atom approach. The concept cluding any previous phase information from the Fourier of using globular sub-units as quasi-atoms was discussed by transform of electron micrographs. The potential distribution David Harker in 1953, when he showed that an appropriate in the projection is assumed a priori to be an assembly of globular scattering factor could be used to normalize the globular densities. By an appropriate dimensional re-scaling, low-resolution diffraction intensities with higher accuracy than these ‘‘globs’’ are then assumed to be pseudo-atoms for the actual atomic scattering factors employed for small mol- normalization of the observed structure factors. After this ecule structures -

Solid State Physics: Problem Set #3 Structural Determination Via X-Ray Scattering Due: Friday Jan
Physics 375 Fall (12) 2003 Solid State Physics: Problem Set #3 Structural Determination via X-Ray Scattering Due: Friday Jan. 31 by 6 pm Reading assignment: for Monday, 3.2-3.3 (structure factors for different lattices) for Wednesday, 3.4-3.7 (scattering methods) for Friday, 4.1-4.3 (mechanical properties of crystals) Problem assignment: Chapter 3 Problems: *3.1 Debye-Scherrer analysis (identify the crystal structure) [Robert] 3.2 Lattice parameter from Debye-Scherrer data 3.11 Measuring thermal expansion with X-ray scattering A1. The unit cell dimension of fcc copper is 0.36 nm. Calculate the longest wavelength of X- rays which will produce diffraction from the close packed planes. From what planes could X- rays with l=0.50 nm be diffracted? *A2. Single crystal diffraction: A cubic crystal with lattice spacing 0.4 nm is mounted with its [001] axis perpendicular to an incident X-ray beam of wavelength 0.1 nm. Initially the crystal is set so as to produce a diffracted beam associated with the (020) planes. Calculate the angle through which the crystal must be turned in order to produce a beam from the (hkl) planes where: a) (hkl)=(030) b) (hkl)=(130) [Brian] c) Which of these diffracted beams would be forbidden if the crystal is: i) sc; ii) bcc; iii) fcc A3. Structure factors for the fcc and diamond bases. a) Construct the structure factor Shkl for the fcc lattice and show that it vanishes unless h, k, and l are all even or all odd. b) Construct the structure factor Shkl for the diamond lattice and show that it vanishes unless h, k, and l are all odd or h+k+l=4n where n is an integer. -
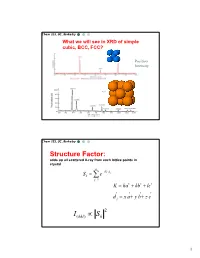
Simple Cubic Lattice
Chem 253, UC, Berkeley What we will see in XRD of simple cubic, BCC, FCC? Position Intensity Chem 253, UC, Berkeley Structure Factor: adds up all scattered X-ray from each lattice points in crystal n iKd j Sk e j1 K ha kb lc d j x a y b z c 2 I(hkl) Sk 1 Chem 253, UC, Berkeley X-ray scattered from each primitive cell interfere constructively when: eiKR 1 2d sin n For n-atom basis: sum up the X-ray scattered from the whole basis Chem 253, UC, Berkeley ' k d k d di R j ' K k k Phase difference: K (di d j ) The amplitude of the two rays differ: eiK(di d j ) 2 Chem 253, UC, Berkeley The amplitude of the rays scattered at d1, d2, d3…. are in the ratios : eiKd j The net ray scattered by the entire cell: n iKd j Sk e j1 2 I(hkl) Sk Chem 253, UC, Berkeley For simple cubic: (0,0,0) iK0 Sk e 1 3 Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis 1 2 iK ( x y z ) iKd j iK0 2 Sk e e e j1 1 ei (hk l) 1 (1)hkl S=2, when h+k+l even S=0, when h+k+l odd, systematical absence Chem 253, UC, Berkeley For BCC: (0,0,0), (1/2, ½, ½)…. Two point basis S=2, when h+k+l even S=0, when h+k+l odd, systematical absence (100): destructive (200): constructive 4 Chem 253, UC, Berkeley Observable diffraction peaks h2 k 2 l 2 Ratio SC: 1,2,3,4,5,6,8,9,10,11,12. -

CHEM 3030 Introduction to X-Ray Crystallography X-Ray Diffraction Is the Premier Technique for the Determination of Molecular
CHEM 3030 Introduction to X-ray Crystallography X-ray diffraction is the premier technique for the determination of molecular structure in chemistry and biochemistry. There are three distinct parts to a structural determination once a high quality single crystal is grown and mounted on the diffractometer. 1. Geometric data collection – the unit cell dimensions are determined from the angles of a few dozen reflections. 2. Intensity Data Collection – the intensity of several thousand reflections are measured. 3. Structure solution – using Direct or Patterson methods, the phase problem is cracked and a function describing the e-density in the unit cell is generated (Fourier synthesis) from the measured intensities. Least squares refinement then optimizes agreement between Fobs (from Intensity data) and Fcalc (from structure). The more tedious aspects of crystallography have been largely automated and the computations are now within the reach of any PC. Crystallography provides an elegant application of symmetry concepts, mathematics (Fourier series), and computer methods to a scientific problem. Crystal structures are now ubiquitous in chemistry and biochemistry. This brief intro is intended to provide the minimum needed to appreciate literature data. FUNDAMENTALS 1. The 7 crystal systems and 14 Bravais lattices. 2. Space group Tables, special and general positions, translational symmetry elements, screw axes and glide planes. 3. Braggs law. Reflections occur only for integral values of the indices hkl because the distance traveled by an X-ray photon through a unit cell must coincide with an integral number of wavelengths. When this condition is satisfied scattering contributions from all unit cells add to give a net scattered wave with intensity I hkl at an angle Θhkl . -

Chapter 3 X-Ray Diffraction • Bragg's Law • Laue's Condition
Chapter 3 X-ray diffraction • Bragg’s law • Laue’s condition • Equivalence of Bragg’s law and Laue’s condition • Ewald construction • geometrical structure factor 1 Bragg’s law Consider a crystal as made out of parallel planes of ions, spaced a distance d apart. The conditions for a sharp peak in the intensity of the scattered radiation are 1. That the x-rays should be specularly reflected by the ions in any one plane and 2. That the reflected rays from successive planes should interfere constructively Path difference between two rays reflected from adjoining planes: 2d sin θ For the rays to interfere constructively, this path difference must be an integral number of wavelength λ nλ = 2d sin θ Bragg’s condition. 2 Bragg angle θ is just the half of the total angle 2 θ by which the incident beam is deflected. There are different ways of sectioning the crystal into planes, each of which will it self produce further reflection. The same portion of Bravais lattice shown in the previous page, with a different way of sectioning the crystal planes. The incident ray is the same. But both the direction and wavelength (determined by Bragg condition with d replaced by d’) of the reflected ray are different from the previous page. 3 Von Laue formulation of X-ray diffraction by a crystal • No particular sectioning of crystal planes r • Regard the crystal as composed of identical microscopic objects placed at Bravais lattice site R • Each of the object at lattice site reradiate the incident radiation in all directions. -

3.014 Materials Laboratory Oct. 13Th – Oct. 20Th , 2006 Lab Week 2 – Module Γ1 Derivative Structures Instructor: Meri Tr
3.014 Materials Laboratory Oct. 13th – Oct. 20th , 2006 Lab Week 2 – Module γ1 Derivative Structures Instructor: Meri Treska OBJECTIVES 9 Review principles of x-ray scattering from crystalline materials 9 Learn how to conduct x-ray powder diffraction experiments and use PDFs 9 Study the inter-relationship of different crystal structures SUMMARY OF TASKS 1) Calculate structure factor for materials to be investigated 2) Prepare samples for x-ray powder diffraction 2) Obtain x-ray scattering patterns for all materials 3) Compare obtained patterns with calculations and powder diffraction files (PDFs) 4) Perform peak fitting to determine percent crystallinity and crystallite size 1 BACKGROUND X-ray Diffraction from Crystalline Materials As discussed in 3.012, a periodic arrangement of atoms will give rise to constructive interference of scattered radiation having a wavelength λ comparable to the periodicity d when Bragg’s law is satisfied: nλ = 2sd inθ where n is an integer and θ is the angle of incidence. Bragg’s law tells us necessary conditions for diffraction, but provides no information regarding peak intensities. To use x-ray diffraction as a tool for materials identification, we must understand the relationship between structure/chemistry and the intensity of diffracted x-rays. Recall from 3.012 class that for a 1d array of atoms, the condition for constructive interference can be determined as follows: unit vector of ur diffracted beam S x ν a µ y ur S 0 x = a cos ν unit vector of ya= cos µ incident beam The total path difference: xy−=a cos ν − acos µλ= h uruurr (SS− 0 )⋅ a=hλ uruur r (SS− 0 ) Defining s = , the condition for 1d constructive interference becomes: λ r r s ⋅ ah= 2 r r For 3 dimensions, we have: s ⋅ ah= r r s ⋅bk= r r s ⋅ cl= where h, k and l are the Miller indices of the scattering plane. -

Section 2: X-Ray Diffraction and Reciprocal Lattice
Physics 927 E.Y.Tsymbal Section 2: X-ray Diffraction and Reciprocal Lattice Bragg law. Most methods for determining the atomic structure of crystals are based of the idea of scattering of radiation. X-rays is one of the types of the radiation which can be used. The wavelength of the radiation should have a wavelength comparable to a typical interatomic distance which is in solids of a few angstroms (10-8 cm). The x-ray wavelength λ can be estimated as follows hc 12.4 Eh==νλ⇒(Å) = . (2.1) λ Ek()eV Therefore, x-rays of energy 2-10 keV are suitable for studying the crystal structure. X-rays interact with electronic shells of atoms in a solid. Electrons absorb and re-radiate x-rays which can then be detected. Nuclei are too heavy to respond. The reflectivity of x-rays is of the order of 10-3- 10-5, so that the penetration in the solid is deep. Therefore, x-rays serve as a bulk probe. In 1913 Bragg found that crystalline solids have remarkably characteristic patterns of reflected x-ray radiation. In crystalline materials, for certain wavelengths and incident directions, intense peaks of scattered radiation were observed. Bragg accounted for this by regarding a crystal as made out of parallel planes of atoms, spaced by distance d apart. The conditions for a sharp peak in the intensity of the scattered radiation were that: (1) the x-rays should be specularly reflected by the atoms in one plane; (2) the reflected rays from the successive planes interfere constructively. -

Crystal Structure Factor Calculations
Crystal Structure factor calculations (A) X-Ray Scattering by an Atom ❑ The conventional UC has lattice points as the vertices ❑ There may or may not be atoms located at the lattice points ❑ The shape of the UC is a parallelepiped in 3D ❑ There may be additional atoms in the UC due to two reasons: ➢ The chosen UC is non-primitive ➢ The additional atoms may be part of the motif Scattering by the Unit cell (uc) ▪ Coherent Scattering ▪ Unit Cell (UC) is representative of the crystal structure ▪ Scattered waves from various atoms in the UC interfere to create the diffraction pattern The wave scattered from the middle plane is out of phase with the ones scattered from top and bottom planes a AC = d = h00 h MCN :: AC :: RBS :: AB :: x AB x x = = AC a h = MCN = 2d Sin( ) = R1R2 h00 2 AB x = = = RBS = = 2 R1R3 AC a h 2 x x = = 2 h x R1R3 → fractional coordinate → x R R = 2 hx a a 1 3 h a Extending to 3D =2 (h x + k y + l z ) Independent of the shape of UC Note: R1 is from corner atoms and R3 is from atoms in additional positions in UC In complex notation =2 (h x + k y + l z ) E== Aei fe i[2 ( h x++ k y l z )] ▪ If atom B is different from atom A → the amplitudes must be weighed by the respective atomic scattering factors (f) ▪ The resultant amplitude of all the waves scattered by all the atoms in the UC gives the scattering factor for the unit cell ▪ The unit cell scattering factor is called the Structure Factor (F) Scattering by an unit cell = f(position of the atoms, atomic scattering factors) Amplitude of wave scattered -

Characterization of Polymer Blends and Block Copolymers by Neutron Scattering
Characterization of Polymer Blends Miscibility, Morphology and Interfaces Mortensen, Kell Published in: Characterization of Polymer Blends and Block Copolymers by Neutron Scattering Publication date: 2014 Document version Early version, also known as pre-print Citation for published version (APA): Mortensen, K. (2014). Characterization of Polymer Blends: Miscibility, Morphology and Interfaces. In Characterization of Polymer Blends and Block Copolymers by Neutron Scattering: Miscibility and Nanoscale Morphology (Wiely-VCH Verlag ed., pp. 237-268). Wiley-VCH. Download date: 26. sep.. 2021 237 7 Characterization of Polymer Blends and Block Copolymers by Neutron Scattering: Miscibility and Nanoscale Morphology Kell Mortensen 7.1 Introduction The interaction between materials and radiation takes a variety of forms, includ- ing absorption and fluorescence, refraction, scattering and reflection. These types of interaction are all tightly related in terms of physical quantities. In this chapter, attention will be focused on the scattering term, when used to determine materi- als’ properties such as miscibility and nanoscale structure. The method relies on the wave-character of the radiation; this is the case whether using electromagnetic beams of light or X-rays with oscillating electric and magnetic fields, or particle radiation such as neutrons or electrons. In the latter cases, it is the de Broglie wave character of the particles that is the relevant quantity. Insight into structural properties using scattering techniques appears as a result of the interference between radiation that is scattered from different sites in the sample. A simple illustration is given in the Young interference experiment, shown in Figure 7.1, where the radiation of plane waves propagate through two slits, making an interference pattern that depend on the separation distance between the two slits, and the wavelength of the radiation. -

Structure Factors (Continued), the Phase Problem, Patterson Techniques and Direct Methods
Materials 286C/UCSB: Class VI — Structure factors (continued), the phase problem, Patterson techniques and direct methods Ram Seshadri ([email protected]) Structure factors The structure factor for a system with many atoms, each with its own form factor fj and sitting within the unit cell at a site with the crystallographic coordinates (uj, vj, wj) is described by: N X Fhkl = fj exp[2πi(huj + kvj + lwj)] j=1 The square of the structure factor is an indication of the intensity of any spot/peak/intensity in the diffraction pattern corresponding to the Bragg conditions being satisfied for the particular hkl. There are other contributors to the intensity, such as the Lorentz-polarization factors and the Debye-Waller factor that we will discuss at a later stage. Some applications: CsCl The CsCl structure is simple cubic (P m3m) with the Cs atom at the corners of a cube at (0,0,0) 1 1 1 and the Cl atom at ( 2 , 2 , 2 ). The structure factor for any hkl reflection is: 1 1 1 F = f exp[2πi(h0 + k0 + l0)] + f exp[2πi(h + k + l )] hkl Cs Cl 2 2 2 which simplifies to Fhkl = fCs + fCl exp[πi(h + k + l)] When (h + k + l) is even, exp[πi(h + k + l)] = 1 and when (h + k + l) is odd, exp[πi(h + k + l)] = −1 This means, for CsCl: ( fCs + fCl, if (h + k + l) is even; Fhkl = fCs − fCl, if (h + k + l) is odd For an hcp metal 1 2 1 The atoms are at (0,0,0) and ( 3 , 3 , 2 ), and both atoms have a form factor f: 1 2 1 F = f + f exp[2πi(h + k + l )] hkl 3 3 2 We consider specific cases: F001 = f + f exp[πi] = f − f = 0 1 F002 = f + f exp[2πi] = f + f = 2f √ 2πi 1 3 F = f + f exp[ ] = f + if 100 3 2 2 √ 1 1 3 3 F = f + f exp[2πi( + )] = f − if 101 3 2 2 2 The corresponding intensities are obtained from ∗ Ihkl = Fhkl · Fhkl Thus 2 2 2 I001 = 0; I002 = 4f ,I100 = f ,I101 = 3f The Argand diagram can be used to graphically represent Fhkl: f F002 F100 ¡ φ 2π 3 F101 ¡ φ 5π 3 2 Diffraction from a centrosymmetric crystal In a centrosymmetric crystal, (u, v, w) and (−u, −v, −w) represent the same point.