Native-Like Secondary Structure in a Peptide From
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Structural Mechanisms of Oligomer and Amyloid Fibril Formation by the Prion Protein Cite This: Chem
ChemComm View Article Online FEATURE ARTICLE View Journal | View Issue Structural mechanisms of oligomer and amyloid fibril formation by the prion protein Cite this: Chem. Commun., 2018, 54,6230 Ishita Sengupta a and Jayant B. Udgaonkar *b Misfolding and aggregation of the prion protein is responsible for multiple neurodegenerative diseases. Works from several laboratories on folding of both the WT and multiple pathogenic mutant variants of the prion protein have identified several structurally dissimilar intermediates, which might be potential precursors to misfolding and aggregation. The misfolded aggregates themselves are morphologically distinct, critically dependent on the solution conditions under which they are prepared, but always b-sheet rich. Despite the lack of an atomic resolution structure of the infectious pathogenic agent in prion diseases, several low resolution models have identified the b-sheet rich core of the aggregates formed in vitro, to lie in the a2–a3 subdomain of the prion protein, albeit with local stabilities that vary Received 17th April 2018, with the type of aggregate. This feature article describes recent advances in the investigation of in vitro Accepted 14th May 2018 prion protein aggregation using multiple spectroscopic probes, with particular focus on (1) identifying DOI: 10.1039/c8cc03053g aggregation-prone conformations of the monomeric protein, (2) conditions which trigger misfolding and oligomerization, (3) the mechanism of misfolding and aggregation, and (4) the structure of the misfolded rsc.li/chemcomm intermediates and final aggregates. 1. Introduction The prion protein can exist in two distinct structural isoforms: a National Centre for Biological Sciences, Tata Institute of Fundamental Research, PrPC and PrPSc. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

DNA Energy Landscapes Via Calorimetric Detection of Microstate Ensembles of Metastable Macrostates and Triplet Repeat Diseases
DNA energy landscapes via calorimetric detection of microstate ensembles of metastable macrostates and triplet repeat diseases Jens Vo¨ lkera, Horst H. Klumpb, and Kenneth J. Breslauera,c,1 aDepartment of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, 610 Taylor Rd, Piscataway, NJ 08854; bDepartment of Molecular and Cell Biology, University of Cape Town, Private Bag, Rondebosch 7800, South Africa; and cCancer Institute of New Jersey, New Brunswick, NJ 08901 Communicated by I. M. Gelfand, Rutgers, The State University of New Jersey, Piscataway, NJ, October 15, 2008 (received for review September 8, 2008) Biopolymers exhibit rough energy landscapes, thereby allowing biological role(s), if any, of kinetically stable (metastable) mi- biological processes to access a broad range of kinetic and ther- crostates that make up the time-averaged, native state ensembles modynamic states. In contrast to proteins, the energy landscapes of macroscopic nucleic acid states remains to be determined. of nucleic acids have been the subject of relatively few experimen- As part of an effort to address this deficiency, we report here tal investigations. In this study, we use calorimetric and spectro- experimental evidence for the presence of discrete microstates scopic observables to detect, resolve, and selectively enrich ener- in metastable triplet repeat bulge looped ⍀-DNAs of potential getically discrete ensembles of microstates within metastable DNA biological significance. The specific triplet repeat bulge looped structures. Our results are consistent with metastable, ‘‘native’’ ⍀-DNA species investigated mimic slipped DNA structures DNA states being composed of an ensemble of discrete and corresponding to intermediates in the processes that lead to kinetically stable microstates of differential stabilities, rather than DNA expansion in triplet repeat diseases (36). -
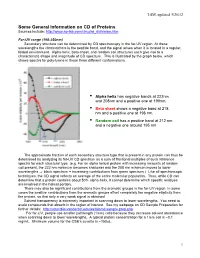
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Conformational Search for the Protein Native State
CHAPTER 19 CONFORMATIONAL SEARCH FOR THE PROTEIN NATIVE STATE Amarda Shehu Assistant Professor Dept. of Comp. Sci. Al. Appnt. Dept. of Comp. Biol. and Bioinf. George Mason University 4400 University Blvd. MSN 4A5 Fairfax, Virginia, 22030, USA Abstract This chapter presents a survey of computational methods that obtain a struc- tural description of the protein native state. This description is important to understand a protein's biological function. The chapter presents the problem of characterizing the native state in conformational detail in terms of the chal- lenges that it raises in computation. Computing the conformations populated by a protein under native conditions is cast as a search problem. Methods such as Molecular Dynamics and Monte Carlo are treated rst. Multiscaling, the combination of reduced and high complexity models of conformations, is briey summarized as a powerful strategy to rapidly extract important fea- tures of the energy surface associated with the protein conformational space. Other strategies that narrow the search space through information obtained in the wet lab are also presented. The chapter then focuses on enhanced sam- Protein Structure Prediction:Method and Algorithms. By H. Rangwala & G. Karypis 1 Copyright c 2013 John Wiley & Sons, Inc. 2 CONFORMATIONAL SEARCH FOR THE PROTEIN NATIVE STATE pling strategies employed to compute native-like conformations when given only amino-acid sequence. Fragment-based assembly methods are analyzed for their success and what they are revealing about the physical process of folding. The chapter concludes with a discussion of future research directions in the computational quest for the protein native state. 19.1 THE QUEST FOR THE PROTEIN NATIVE STATE From the rst formulation of the protein folding problem by Wu in 1931 to the experiments of Mirsky and Pauling in 1936, chemical and physical properties of protein molecules were attributed to the amino-acid composition and structural arrangement of the protein chain [86, 56]. -

Protein Folding and Structure Prediction
Protein Folding and Structure Prediction A Statistician's View Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Proteins Amino acids without peptide bonds. Amino acids with peptide bonds. ¡ Amino acids are the building blocks of proteins. Proteins Both figures show the same protein (the bacterial protein L). The right figure also highlights the secondary structure elements. Space Resolution limit of a light microscope Glucose Ribosome Red blood cell C−C bond Hemoglobin Bacterium 1 10 100 1000 10000 100000 1nm 1µm Distance [ A° ] Energy C−C bond Green Noncovalent bond light Glucose Thermal ATP 0.1 1 10 100 1000 Energy [ kcal/mol ] Non-Bonding Interactions Amino acids of a protein are joined by covalent bonding interactions. The polypep- tide is folded in three dimension by non-bonding interactions. These interactions, which can easily be disrupted by extreme pH, temperature, pressure, and denatu- rants, are: Electrostatic Interactions (5 kcal/mol) Hydrogen-bond Interactions (3-7 kcal/mol) Van Der Waals Interactions (1 kcal/mol) ¡ Hydrophobic Interactions ( 10 kcal/mol) The total inter-atomic force acting between two atoms is the sum of all the forces they exert on each other. Energy Profile Transition State Denatured State Native State Radius of Gyration of Denatured Proteins Do chemically denatured proteins behave as random coils? The radius of gyration Rg of a protein is defined as the root mean square dis- tance from each atom of the protein to their centroid. For an ideal (infinitely thin) random-coil chain in a solvent, the average radius 0.5 ¡ of gyration of a random coil is a simple function of its length n: Rg n For an excluded volume polymer (a polymer with non-zero thickness and non- trivial interactions between monomers) in a solvent, the average radius of gyra- 0.588 tion, we have Rg n (Flory 1953). -

A Comparison of the Folding Kinetics of a Small, Artificially Selected DNA Aptamer with Those of Equivalently Simple Naturally Occurring Proteins
A comparison of the folding kinetics of a small, artificially selected DNA aptamer with those of equivalently simple naturally occurring proteins Camille Lawrence,1 Alexis Vallee-B elisle, 2,3 Shawn H. Pfeil,4,5 Derek de Mornay,2 Everett A. Lipman,1,4 and Kevin W. Plaxco1,2* 1Interdepartmental Program in Biomolecular Science and Engineering, University of California, Santa Barbara, California 93106 2Department of Chemistry and Biochemistry, University of California, Santa Barbara, California 93106 3Laboratory of Biosensors and Nanomachines, Departement de Chimie, Universite de Montreal, Quebec, Canada 4Department of Physics, University of California, Santa Barbara, California 93106 5Department of Physics, West Chester University of Pennsylvania, West Chester, Pennsylvania 19383 Received 16 July 2013; Revised 17 October 2013; Accepted 23 October 2013 DOI: 10.1002/pro.2390 Published online 29 October 2013 proteinscience.org Abstract: The folding of larger proteins generally differs from the folding of similarly large nucleic acids in the number and stability of the intermediates involved. To date, however, no similar com- parison has been made between the folding of smaller proteins, which typically fold without well- populated intermediates, and the folding of small, simple nucleic acids. In response, in this study, we compare the folding of a 38-base DNA aptamer with the folding of a set of equivalently simple proteins. We find that, as is true for the large majority of simple, single domain proteins, the aptamer folds through a concerted, millisecond-scale process lacking well-populated intermedi- ates. Perhaps surprisingly, the observed folding rate falls within error of a previously described relationship between the folding kinetics of single-domain proteins and their native state topology. -

Study of the Variability of the Native Protein Structure
Study of the Variability of the Native Protein Structure Xusi Han, Woong-Hee Shin, Charles W Christoffer, Genki Terashi, Lyman Monroe, and Daisuke Kihara, Purdue University, West Lafayette, IN, United States r 2018 Elsevier Inc. All rights reserved. Introduction Proteins are flexible molecules. After being translated from a messenger RNA by a ribosome, a protein folds into its native structure (the structure of lowest free energy), which is suitable for carrying out its biological function. Although the native structure of a protein is stabilized by physical interactions of atoms including hydrogen bonds, disulfide bonds between cysteine residues, van der Waals interactions, electrostatic interactions, and solvation (interactions with solvent), the structure still admits flexible motions. Motions include those of side-chains, and some parts of main-chains, especially regions that do not form the secondary structures, which are often called loop regions. In many cases, the flexibility of proteins plays an important or essential role in the biological functions of the proteins. For example, for some enzymes, such as triosephosphate isomerase (Derreumaux and Schlick, 1998), loop regions that exist in vicinity of active (i.e., enzymatic reaction) sites, takes part in binding and holding a ligand molecule. Transporters, such as maltose transporter (Chen, 2013), are known to make large open-close motions to transfer ligand molecules across the cellular membrane. For many motor proteins, such as myosin V that “walk” along actin filament as observed in muscle contraction (Kodera and Ando, 2014), flexibility is the central for their functions. Reflecting such intrinsic flexibility of protein structures, differences are observed in protein tertiary structures determined by experimental methods such as X-ray crystallography, nuclear magnetic resonance (NMR) when they are solved under different conditions. -

Evolution of Biomolecular Structure Class II Trna-Synthetases and Trna
University of Illinois at Urbana-Champaign Luthey-Schulten Group Theoretical and Computational Biophysics Group Evolution of Biomolecular Structure Class II tRNA-Synthetases and tRNA MultiSeq Developers: Prof. Zan Luthey-Schulten Elijah Roberts Patrick O’Donoghue John Eargle Anurag Sethi Dan Wright Brijeet Dhaliwal March 2006. A current version of this tutorial is available at http://www.ks.uiuc.edu/Training/Tutorials/ CONTENTS 2 Contents 1 Introduction 4 1.1 The MultiSeq Bioinformatic Analysis Environment . 4 1.2 Aminoacyl-tRNA Synthetases: Role in translation . 4 1.3 Getting Started . 7 1.3.1 Requirements . 7 1.3.2 Copying the tutorial files . 7 1.3.3 Configuring MultiSeq . 8 1.3.4 Configuring BLAST for MultiSeq . 10 1.4 The Aspartyl-tRNA Synthetase/tRNA Complex . 12 1.4.1 Loading the structure into MultiSeq . 12 1.4.2 Selecting and highlighting residues . 13 1.4.3 Domain organization of the synthetase . 14 1.4.4 Nearest neighbor contacts . 14 2 Evolutionary Analysis of AARS Structures 17 2.1 Loading Molecules . 17 2.2 Multiple Structure Alignments . 18 2.3 Structural Conservation Measure: Qres . 19 2.4 Structure Based Phylogenetic Analysis . 21 2.4.1 Limitations of sequence data . 21 2.4.2 Structural metrics look further back in time . 23 3 Complete Evolutionary Profile of AspRS 26 3.1 BLASTing Sequence Databases . 26 3.1.1 Importing the archaeal sequences . 26 3.1.2 Now the other domains of life . 27 3.2 Organizing Your Data . 28 3.3 Finding a Structural Domain in a Sequence . 29 3.4 Aligning to a Structural Profile using ClustalW .