Molecular Dynamics of Folded and Disordered Polypeptides in Comparison with Nuclear
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Using Constrained Density Functional Theory to Track Proton Transfers and to Sample Their Associated Free Energy Surface
Using Constrained Density Functional Theory to Track Proton Transfers and to Sample Their Associated Free Energy Surface Chenghan Li and Gregory A. Voth* Department of Chemistry, Chicago Center for Theoretical Chemistry, James Franck Institute, and Institute for Biophysical Dynamics, University of Chicago, Chicago, IL, 60637 Keywords: free energy sampling, proton transport, density functional theory, proton transfer ABSTRACT: The ab initio molecular dynamics (AIMD) and quantum mechanics/molecular mechanics (QM/MM) methods are powerful tools for studying proton solvation, transfer, and transport processes in various environments. However, due to the high computational cost of such methods, achieving sufficient sampling of rare events involving excess proton motion – especially when Grotthuss proton shuttling is involved – usually requires enhanced free energy sampling methods to obtain informative results. Moreover, an appropriate collective variable (CV) that describes the effective position of the net positive charge defect associated with an excess proton is essential for both tracking the trajectory of the defect and for the free energy sampling of the processes associated with the resulting proton transfer and transport. In this work, such a CV is derived from first principles using constrained density functional theory (CDFT). This CV is applicable to a broad array of proton transport and transfer processes as studied via AIMD and QM/MM simulation. 1 INTRODUCTION The accurate and efficient delineation of proton transport (PT) and -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development
processes Review Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development Outi M. H. Salo-Ahen 1,2,* , Ida Alanko 1,2, Rajendra Bhadane 1,2 , Alexandre M. J. J. Bonvin 3,* , Rodrigo Vargas Honorato 3, Shakhawath Hossain 4 , André H. Juffer 5 , Aleksei Kabedev 4, Maija Lahtela-Kakkonen 6, Anders Støttrup Larsen 7, Eveline Lescrinier 8 , Parthiban Marimuthu 1,2 , Muhammad Usman Mirza 8 , Ghulam Mustafa 9, Ariane Nunes-Alves 10,11,* , Tatu Pantsar 6,12, Atefeh Saadabadi 1,2 , Kalaimathy Singaravelu 13 and Michiel Vanmeert 8 1 Pharmaceutical Sciences Laboratory (Pharmacy), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland; ida.alanko@abo.fi (I.A.); rajendra.bhadane@abo.fi (R.B.); parthiban.marimuthu@abo.fi (P.M.); atefeh.saadabadi@abo.fi (A.S.) 2 Structural Bioinformatics Laboratory (Biochemistry), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland 3 Faculty of Science-Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CH Utrecht, The Netherlands; [email protected] 4 Swedish Drug Delivery Forum (SDDF), Department of Pharmacy, Uppsala Biomedical Center, Uppsala University, 751 23 Uppsala, Sweden; [email protected] (S.H.); [email protected] (A.K.) 5 Biocenter Oulu & Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7 A, FI-90014 Oulu, Finland; andre.juffer@oulu.fi 6 School of Pharmacy, University of Eastern Finland, FI-70210 Kuopio, Finland; maija.lahtela-kakkonen@uef.fi (M.L.-K.); tatu.pantsar@uef.fi -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

Molecular Dynamics Study of the Stress–Strain Behavior of Carbon-Nanotube Reinforced Epon 862 Composites R
Materials Science and Engineering A 447 (2007) 51–57 Molecular dynamics study of the stress–strain behavior of carbon-nanotube reinforced Epon 862 composites R. Zhu a,E.Pana,∗, A.K. Roy b a Department of Civil Engineering, University of Akron, Akron, OH 44325, USA b Materials and Manufacturing Directorate, Air Force Research Laboratory, AFRL/MLBC, Wright-Patterson Air Force Base, OH 45433, USA Received 9 March 2006; received in revised form 2 August 2006; accepted 20 October 2006 Abstract Single-walled carbon nanotubes (CNTs) are used to reinforce epoxy Epon 862 matrix. Three periodic systems – a long CNT-reinforced Epon 862 composite, a short CNT-reinforced Epon 862 composite, and the Epon 862 matrix itself – are studied using the molecular dynamics. The stress–strain relations and the elastic Young’s moduli along the longitudinal direction (parallel to CNT) are simulated with the results being also compared to those from the rule-of-mixture. Our results show that, with increasing strain in the longitudinal direction, the Young’s modulus of CNT increases whilst that of the Epon 862 composite or matrix decreases. Furthermore, a long CNT can greatly improve the Young’s modulus of the Epon 862 composite (about 10 times stiffer), which is also consistent with the prediction based on the rule-of-mixture at low strain level. Even a short CNT can also enhance the Young’s modulus of the Epon 862 composite, with an increment of 20% being observed as compared to that of the Epon 862 matrix. © 2006 Elsevier B.V. All rights reserved. Keywords: Carbon nanotube; Epon 862; Nanocomposite; Molecular dynamics; Stress–strain curve 1. -
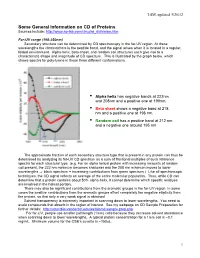
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

FORCE FIELDS for PROTEIN SIMULATIONS by JAY W. PONDER
FORCE FIELDS FOR PROTEIN SIMULATIONS By JAY W. PONDER* AND DAVIDA. CASEt *Department of Biochemistry and Molecular Biophysics, Washington University School of Medicine, 51. Louis, Missouri 63110, and tDepartment of Molecular Biology, The Scripps Research Institute, La Jolla, California 92037 I. Introduction. ...... .... ... .. ... .... .. .. ........ .. .... .... ........ ........ ..... .... 27 II. Protein Force Fields, 1980 to the Present.............................................. 30 A. The Am.ber Force Fields.............................................................. 30 B. The CHARMM Force Fields ..., ......... 35 C. The OPLS Force Fields............................................................... 38 D. Other Protein Force Fields ....... 39 E. Comparisons Am.ong Protein Force Fields ,... 41 III. Beyond Fixed Atomic Point-Charge Electrostatics.................................... 45 A. Limitations of Fixed Atomic Point-Charges ........ 46 B. Flexible Models for Static Charge Distributions.................................. 48 C. Including Environmental Effects via Polarization................................ 50 D. Consistent Treatment of Electrostatics............................................. 52 E. Current Status of Polarizable Force Fields........................................ 57 IV. Modeling the Solvent Environment .... 62 A. Explicit Water Models ....... 62 B. Continuum Solvent Models.......................................................... 64 C. Molecular Dynamics Simulations with the Generalized Born Model........ -

Force Fields for MD Simulations
Force Fields for MD simulations • Topology/parameter files • Where do the numbers an MD code uses come from? • How to make topology files for ligands, cofactors, special amino acids, … • How to obtain/develop missing parameters. • QM and QM/MM force fields/potential energy descriptions used for molecular simulations. The Potential Energy Function Ubond = oscillations about the equilibrium bond length Uangle = oscillations of 3 atoms about an equilibrium bond angle Udihedral = torsional rotation of 4 atoms about a central bond Unonbond = non-bonded energy terms (electrostatics and Lenard-Jones) Energy Terms Described in the CHARMm Force Field Bond Angle Dihedral Improper Classical Molecular Dynamics r(t +!t) = r(t) + v(t)!t v(t +!t) = v(t) + a(t)!t a(t) = F(t)/ m d F = ! U (r) dr Classical Molecular Dynamics 12 6 &, R ) , R ) # U (r) = . $* min,ij ' - 2* min,ij ' ! 1 qiq j ij * ' * ' U (r) = $ rij rij ! %+ ( + ( " 4!"0 rij Coulomb interaction van der Waals interaction Classical Molecular Dynamics Classical Molecular Dynamics Bond definitions, atom types, atom names, parameters, …. What is a Force Field? In molecular dynamics a molecule is described as a series of charged points (atoms) linked by springs (bonds). To describe the time evolution of bond lengths, bond angles and torsions, also the non-bonding van der Waals and elecrostatic interactions between atoms, one uses a force field. The force field is a collection of equations and associated constants designed to reproduce molecular geometry and selected properties of tested structures. Energy Functions Ubond = oscillations about the equilibrium bond length Uangle = oscillations of 3 atoms about an equilibrium bond angle Udihedral = torsional rotation of 4 atoms about a central bond Unonbond = non-bonded energy terms (electrostatics and Lenard-Jones) Parameter optimization of the CHARMM Force Field Based on the protocol established by Alexander D. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

The Fip35 WW Domain Folds with Structural and Mechanistic Heterogeneity in Molecular Dynamics Simulations
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Biophysical Journal Volume 96 April 2009 L53–L55 L53 The Fip35 WW Domain Folds with Structural and Mechanistic Heterogeneity in Molecular Dynamics Simulations Daniel L. Ensign and Vijay S. Pande* Department of Chemistry, Stanford University, Stanford, California ABSTRACT We describe molecular dynamics simulations resulting in the folding the Fip35 Hpin1 WW domain. The simulations were run on a distributed set of graphics processors, which are capable of providing up to two orders of magnitude faster compu- tation than conventional processors. Using the Folding@home distributed computing system, we generated thousands of inde- pendent trajectories in an implicit solvent model, totaling over 2.73 ms of simulations. A small number of these trajectories folded; the folding proceeded along several distinct routes and the system folded into two distinct three-stranded b-sheet conformations, showing that the folding mechanism of this system is distinctly heterogeneous. Received for publication 8 September 2008 and in final form 22 January 2009. *Correspondence: [email protected] Because b-sheets are a ubiquitous protein structural motif, tories on the distributed computing environment, Folding@ understanding how they fold is imperative in solving the home (5). For these calculations, we utilized graphics pro- protein folding problem. Liu et al. (1) recently made a heroic cessing units (ATI Technologies; Sunnyvale, CA), deployed set of measurements of the folding kinetics of 35 three- by the Folding@home contributors. Using optimized code, stranded b-sheet sequences derived from the Hpin1 WW individual graphics processing units (GPUs) of the type domain. -

Computational Biophysics: Introduction
Computational Biophysics: Introduction Bert de Groot, Jochen Hub, Helmut Grubmüller Max Planck-Institut für biophysikalische Chemie Theoretische und Computergestützte Biophysik Am Fassberg 11 37077 Göttingen Tel.: 201-2308 / 2314 / 2301 / 2300 (Secr.) Email: [email protected] [email protected] [email protected] www.mpibpc.mpg.de/grubmueller/ Chloroplasten, Tylakoid-Membran From: X. Hu et al., PNAS 95 (1998) 5935 Primary steps in photosynthesis F-ATP Synthase 20 nm F1-ATP(synth)ase ATP hydrolysis drives rotation of γ subunit and attached actin filament F1-ATP(synth)ase NO INERTIA! Proteins are Molecular Nano-Machines ! Elementary steps: Conformational motions Overview: Computational Biophysics: Introduction L1/P1: Introduction, protein structure and function, molecular dynamics, approximations, numerical integration, argon L2/P2: Tertiary structure, force field contributions, efficient algorithms, electrostatics methods, protonation, periodic boundaries, solvent, ions, NVT/NPT ensembles, analysis L3/P3: Protein data bank, structure determination by NMR / x-ray; refinement L4/P4: Monte Carlo, normal mode analysis, principal components L5/P5: Bioinformatics: sequence alignment, Structure prediction, homology modelling L6/P6: Charge transfer & photosynthesis, electrostatics methods L7/P7: Aquaporin / ATPase: two examples from current research Overview: Computational Biophysics: Concepts & Methods L08/P08: MD Simulation & Markov Theory: Molecular Machines L09/P09: Free energy calculations: Molecular recognition L10/P10: Non-equilibrium thermodynamics: