POLYMER STRUCTURE and CHARACTERIZATION Professor
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Polymers Division
National Institute of Standards and Technology Technology Administration U.S. Department of MSEL Commerce NISTIR 6796 FY 2001 PROGRAMS September 2001 AND ACCOMPLISHMENTS POLYMERS DIVISION Advance measurement methods to characterize microstructure failure in composites This optical image is a planar array of glass fibers embedded in epoxy resin matrix deformed under tension. The arrays are used to investigate the failure initiation in fibrous composites. In the optical image, the fibers are spaced (30 to 45) mm apart and the dark regions along each fiber are cracks that extend into the surrounding polymer matrix. The light blue regions that connect the fiber breaks show high deformation shear bands that emanate from the tips of the matrix cracks. National Institute of Standards and Technology Karen H. Brown, MATERIALS Acting Director Technology SCIENCE AND Administration ENGINEERING U.S. Department of Commerce Donald L. Evans, Secretary LABORATORY T OF C EN OM M M T E R R FY 2001 PROGRAMS A C P E E D AND U N A I C T I E R D E S M ACCOMPLISHMENTS T A ATES OF POLYMERS DIVISION Eric J. Amis, Chief Bruno M. Fanconi, Deputy NISTIR 6796 September 2001 Table of Contents Executive Summary ............................................................................................................................................................. 1 Technical Highlights Mass Spectrometry of Polyethylene ......................................................................................................................... 4 Combinatorial Study of -

Beyond GPC Light Scattering for Absolute Polymer
J U N E 2 0 1 6 BEYOND GPC: USING LIGHT SCATTERING FOR ABSOLUTE POLYMER CHARACTERIZATION BEYOND GPC: USING LIGHT TOC SCATTERING FOR ABSOLUTE Table of contents POLYMER CHARACTERIZation Adding MALS Detection to GPC Overcoming Fear, Uncertainty, and Doubt in GPC: The Need for an Absolute Measurement of Molar Mass 04 Mark W. Spears, Jr. Characterizing Polymer Branching Principles of Detection and Characterization of Branching in Synthetic and Natural Polymers by MALS 11 Stepan Podzimek Analyzing Polymerization Processes Light-Scattering Techniques for Analyzing Polymerization Processes An interview with Judit E. Puskas 18 SEC–MALS vs. AF4–MALS Characterization of Styrene-Butadiene Rubbers by SEC–MALS and AF4–MALS 20 Stepan Podzimek The Most Interesting Man in Light Scattering. photo: © PeteBleyer.com We Call Him Dad. Dr. Philip Wyatt is the father of Multi-Angle Light delight them with unexpectedly attentive cus- Scattering (MALS) detection. Together with his tomer service. Check. After all, we don’t just want sons, Geof and Cliff, he leads his company to to sell our instruments, we want to help you do produce the industry’s most advanced instruments great work with them. Because at Wyatt Technol- by upholding two core premises: First, build top ogy, our family extends beyond our last name to quality instruments to serve scientists. Check. Then everyone who uses our products. For essential macromolecular and nanoparticle characterization—The Solution is Light™ © 2015 Wyatt Technology. All rights reserved. All trademarks and registered trademarks are properties of their respective holders. OVERCOMING FEAR, UNCERTAINTY, AND DOubT IN GPC: THE NEED FOR AN AbSOluTE MEASUREMENT OF MOLAR MASS Mark W. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

Polymers, Elastomers and Composites SES Offers a Full Range of R&D, Failure Analysis and Analytical Characterization Support Services
Materials Science & Engineering: Polymers, Elastomers and Composites SES offers a full range of R&D, failure analysis and analytical characterization support services. New Facilities-New Capabilities A Full Suite of Thermal Analysis Testing The use of polymers, Instruments elastomers and The backbone of a polymer characterization laboratory composites is growing is a suite of thermal analysis testing instruments. fast. This growth is not SES has a number of research-quality TA Instruments limited to the durable testing equipment including TGA (Thermogravimetric and non-durable Analysis), DSC (Differential Scanning Calorimeter), consumer and medical DMA (Dynamic Mechanical Analysis) and FTIR (Fourier products industries, but Transform, Infrared Spectroscopy). In addition to also includes diverse the ability to fully characterize polymers, polymer industries such as composites and elastomers, SES can conduct manufacturing equipment components, industrial exposure–based aging investigations as a function products, oilfield equipment and pipeline repair, to of applied stress/strain, temperature and time. name a few. Standard exposure-based tests can include about any combination of sustained and variable stress/strain in To meet the growing demand for polymer material contact with operating fluid, explosive decompression selection, failure analysis and material compatibility and high pressure-high temperature (HPHT) aging in assessment services, SES has invested heavily corrosive media, sterilization, physical aging, ultraviolet in staffing and polymer characterization/testing weathering, and simulated life cycle testing. instruments. A new polymer materials services facility has been constructed in SES’s centrally located Ohio facility to meet the TGA (Thermogravimetric Analysis) growing demand The TGA is generally for this work. used for compositional The labs include analysis, determination a broad range of decomposition of equipment temperature and necessary to the development of evaluate the very kinetic models for complex polymer- decomposition. -
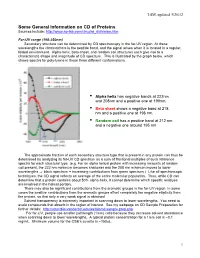
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

HT-GPC for Advanced Polymer Characterization Solution Card
HT-GPC for Advanced Polymer Characterization TechnologyLab High Temperature GPC determines the molecular Customizable reports according to the client’s weight and molecular weight distribution of needs: Mw, Mn, Mz, PDI, SCB/1000C, LCB, polymers soluble only at high temperature, such as implementing calculation models, etc. polyolefins (HDPE, PP, LDPE, LLDPE, copolymers EP, EVA and EBA. Dual detection system with RI and Viscometer which determine the copolymer concentration and its distribution (SCB), as well the molecular structure of polymers (lineal or branched chains and branching distribution, LCB). ● High-sensitive RI detector designed to measure polymer concentration and short chain branching with excellent stability. RI detector incorporates interference filters at five different wavelengths and a thermoelectrically cooled MCT detector with high sensitivity. ● The high temperature differential viscometer detector provides a measurement of intrinsic viscosity and allows the determination of molecular size GPC-IR HT and branching structure. Technology Lab has extensive equipment that allows comprehensive and accurate polymer characterization: ● Chemical characterization by RMN, Raman and FTIR ● Rheological characterization by ARES, Brookfield and Haake ● Structural characterization by GPC, TREF, CRYSTAF ● Morphological characterization by SEM Report of results and MOP-Raman HT-GPC for Advanced Polymer Characterization ● No less than 50mg of sample is needed for ● Chemically cross-linked polymers. polymer characterization by HT-GPC. ● Polymer solubility in the working range of ● Chemically uncrossed material and the HT-GPC (30oC to 220oC). soluble in TCB (trichlorobenzen) at 150 ºC. ● Carbon black must be previously eliminated by filtration from the sample. Gel permeation chromatography (GPC) is a separation technique, commonly used for polymer characterization, which allows differentiate compounds according to their molecular size. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Protein Folding and Structure Prediction
Protein Folding and Structure Prediction A Statistician's View Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Proteins Amino acids without peptide bonds. Amino acids with peptide bonds. ¡ Amino acids are the building blocks of proteins. Proteins Both figures show the same protein (the bacterial protein L). The right figure also highlights the secondary structure elements. Space Resolution limit of a light microscope Glucose Ribosome Red blood cell C−C bond Hemoglobin Bacterium 1 10 100 1000 10000 100000 1nm 1µm Distance [ A° ] Energy C−C bond Green Noncovalent bond light Glucose Thermal ATP 0.1 1 10 100 1000 Energy [ kcal/mol ] Non-Bonding Interactions Amino acids of a protein are joined by covalent bonding interactions. The polypep- tide is folded in three dimension by non-bonding interactions. These interactions, which can easily be disrupted by extreme pH, temperature, pressure, and denatu- rants, are: Electrostatic Interactions (5 kcal/mol) Hydrogen-bond Interactions (3-7 kcal/mol) Van Der Waals Interactions (1 kcal/mol) ¡ Hydrophobic Interactions ( 10 kcal/mol) The total inter-atomic force acting between two atoms is the sum of all the forces they exert on each other. Energy Profile Transition State Denatured State Native State Radius of Gyration of Denatured Proteins Do chemically denatured proteins behave as random coils? The radius of gyration Rg of a protein is defined as the root mean square dis- tance from each atom of the protein to their centroid. For an ideal (infinitely thin) random-coil chain in a solvent, the average radius 0.5 ¡ of gyration of a random coil is a simple function of its length n: Rg n For an excluded volume polymer (a polymer with non-zero thickness and non- trivial interactions between monomers) in a solvent, the average radius of gyra- 0.588 tion, we have Rg n (Flory 1953). -

Helix-Coil Conformational Change Accompanied by Anisotropic–Isotropic Transition
Polymer Journal, Vol.33, No. 11, pp 898—901 (2001) NOTES Helix-Coil Conformational Change Accompanied by Anisotropic–Isotropic Transition † Ning LIU, Jiaping LIN, Tao C HEN, Jianding CHEN, Dafei ZHOU, and Le LI Department of Polymer Science and Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China (Received November 22, 2000; Accepted July 1, 2001) KEY WORDS Helix-Coil Transition / Liquid Crystal Polymer / Polypeptide / Anisotropic– Isotropic Transition / Polypeptides, particularly poly(γ-benzyl-L- limited study of this acid induced anisotropic–isotropic glutamate) (PBLG) and other glutamic acid ester transition has been reported.13, 14 The precise molecular polymers, have the ability to exist in α-helix, a well de- mechanism of the transition remains to be determined. fined chain conformation of long chain order and retain In the present work, the texture changes in the acid- such a structure in numerous organic solvents that sup- induced anisotropic–isotropic transition were studied port intramolecular hydrogen bonding.1, 2 If PBLG and by microscope observations and the corresponding related polypeptides are dissolved in a binary solvent molecular conformational changes were followed by IR mixture containing a non-helicogenic component such measurements. The effect of the polymer concentra- as dichloroacetic acid (DCA) or trifluoroacetic acid tion was studied. According to the information gained (TFA), they can undergo a conformational change from through the experiment, the molecular mechanism with the α-helix to random coil form due to the changes respect to the acid-induced helix-coil conformational in solvent composition, temperature or both.3, 4 The change was suggested. -

Ultrasonic Dynamic Mechanical Analysis of Polymers
Applied_Rheology_Vol_15_5.qxd 051109 13:40 Uhr Seite 326 Ultrasonic Dynamic Mechanical Analysis of Polymers Francesca Lionetto*, Francesco Montagna, Alfonso Maffezzoli Department of Innovation Engineering, University of Lecce, 73100 Lecce, Italy * Email: [email protected] Fax: x39.0832.297525 Received: 18.7.2005, Final version: 23.9.2005 Abstract: The propagation of ultrasonic waves in polymers depends on their viscoelastic behaviour and density, resulting significantly affected by phase transitions occurring with changing temperature and pressure or during chem- ical reactions. Therefore, the application of low intensity ultrasound, acting as a high frequency dynamic mechan- ical deformation applied to a polymer, can monitor the changes of viscoelastic properties associated with the glass transition, the crystallization, the physical or chemical gelation, the crosslinking. Thanks to the non-destruc- tive character (due to the very small deformation amplitude), low intensity ultrasound can be successfully used for polymer characterization. Moreover, this technique has a big potential as a sensor for on-line and in-situ monitoring of production processes for polymers or polymer matrix composites. Recently, in the laboratory of Polymeric Materials of Lecce University a custom made ultrasonic set-up for the characterization of polymeric material, even at high temperatures, has been developed. The ultrasonic equipment is coupled with a rotation- al rheometer. Ultrasonic waves and shear oscillations at low frequency can be applied simultaneously on the sample, getting information on its viscoelastic behaviour over a wide frequency range. The aim of this paper is to present the potential and reliability of the ultrasonic equipment for the ultrasonic dynamic mechanical analy- sis (UDMA) of both thermosetting and thermoplastic polymers. -

Polymer Analysis Chapter 1
Polymer Analysis Chapter 1. Introduction/Overview The focus of this course is analysis and characterization of polymers and plastics. Analysis of polymeric systems is essentially a subtopic of the field of chemical analysis of organic materials. Because of this, spectroscopic techniques commonly used by organic chemists are at the heart of Polymer Analysis, e.g. infra-red (IR) spectroscopy, Raman spectroscopy, nuclear magnetic resonance (NMR) spectroscopy and to some extent ultra-violet/visible (UV/Vis) spectroscopy. In addition, since most polymeric materials are used in the solid state, traditional characterization techniques aimed at the solid state are often encountered, x-ray diffraction, optical and electron microscopy as well as thermal analysis. Unique to polymeric materials are analytic techniques which focus on viscoelastic properties, specifically, dynamic mechanical testing. Additionally, techniques aimed at determination of colloidal scale structure such as chain structure and molecular weight for high molecular weight materials are somewhat unique to polymeric materials, i.e. gel permeation chromatography, small angle scattering (SAS) and various other techniques for the determination of colloidal scale structure. The textbook, Polymer Characterization covers all of these analytic techniques and can serve as a reference for a general introduction to the analysis of polymeric systems. Due to time constraints we can only cover a small number of analytic techniques important to polymers in this course and these are outlined in the syllabus. Structure/Processing/Property: Generally people resort to analytic techniques when confronted with a problem which involves understanding the relationship between properties of a processed material and the structure and chemical composition of the system.