(12) Patent Application Publication (10) Pub. No.: US 2015/0175532 A1 Mudduluru Et Al
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

SAFETY DATA SHEET Revision Date 28.07.2021 According to Regulation (EC) No
Version 7.0 SAFETY DATA SHEET Revision Date 28.07.2021 according to Regulation (EC) No. 1907/2006 Print Date 02.10.2021 GENERIC EU MSDS - NO COUNTRY SPECIFIC DATA - NO OEL DATA SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifiers Product name : Aluminum iodide Product Number : 208493 Brand : Aldrich REACH No. : A registration number is not available for this substance as the substance or its uses are exempted from registration, the annual tonnage does not require a registration or the registration is envisaged for a later registration deadline. CAS-No. : 7784-23-8 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Laboratory chemicals, Manufacture of substances 1.3 Details of the supplier of the safety data sheet Company : Sigma-Aldrich Inc. 3050 SPRUCE ST ST. LOUIS MO 63103 UNITED STATES Telephone : +1 314 771-5765 Fax : +1 800 325-5052 1.4 Emergency telephone Emergency Phone # : 800-424-9300 CHEMTREC (USA) +1-703- 527-3887 CHEMTREC (International) 24 Hours/day; 7 Days/week SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 Skin corrosion (Sub-category 1B), H314 Serious eye damage (Category 1), H318 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 Label elements Labelling according Regulation (EC) No 1272/2008 Aldrich- 208493 Page 1 of 8 The life science business of Merck operates as MilliporeSigma in the US and Canada Pictogram Signal word Danger Hazard statement(s) H314 Causes severe skin burns and eye damage. -
Formula & Equation Writing
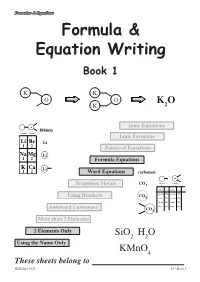
Formulae & Equations Formula & Equation Writing Book 1 K K O O K O K 2 Ionic Equations lithium column column 1 2 Ionic Formulae Li Be Li 1 2 Balanced Equations Na Mg Li 1 2 Formula Equations K Ca Li 1 2 Word Equations carbonate valency valency Transition Metals CO3 1 2 name formula name formula ammonium NH4 carbonate CO3 Using Brackets CO 3 cyanide CN chromate CrO4 hydroxide OH sulphate SO4 nitrate NO sulphite SO Awkward Customers 3 3 CO3 More than 2 Elements 2 Elements Only SiO2 H2O Using the Name Only KMnO4 These sheets belong to KHS Sept 2013 page 1 S3 - Book 1 Formulae & Equations What is a Formula ? The formula of a compound tells you two things about the compound :- SiO H O i) which elements are in the compound 2 2 using symbols, and KMnO4 ii) how many atoms of each element are in the compound using numbers. Test Yourself What would be the formula for each of the following? Br H Cl K H C S Al Br O H Cl K H Br Using the Name Only Some compounds have extra information in their carbon monoxide names that allow people to work out and write CO the correct formula. carbon dioxide CO2 The names of the elements appear as usual but dinitrogen tetroxide N O this time the number of each type of atom is 2 4 included using mono- = 1 di- = 12 tri- = 3 tetra- = 4 penta- = 5 hexa- =46 Test Yourself 1 What would be the formula for each of the following? 1. -

Journal-2019-06
INTELLECTUAL PROPERTY REGISTRY OF SAMOA (IPROS) JOURNAL 2019/06 Publication Date – 4 December 2019 GENERAL INFORMATION The Public is hereby notified that the following persons have applied for the Registration of the Trade Marks as set out in Section 50 of the Intellectual Property Act 2011. Any person who wishes to oppose the registration of any of the under-mentioned Trade Marks, must give notice to the Registrar of Trade Marks, c/- Ministry of Commerce, Industry and Labour, Apia, Samoa within three (3) months from the date of this publication. The notice must be in writing and must include a statement of the grounds of opposition. Each notice of opposition must be accompanied by a filing fee of WST$300. Any person who has any doubts as to whether he/she may be affected by the registration of the Trade Mark in Samoa should consult his/her Solicitor immediately. For any enquiries regarding Trade Marks, please contact the Registry of Intellectual Property at the following telephone number (+685) 20441 or email [email protected] Faafetai, Registrar of Intellectual Property Accepted Trade Marks – Direct Filing (NEWSPAPER VERSION) Application No. : WS/T/7350 Representation of Mark Filing Date : 12 September, 2018 Priority Date : N/A Proprietor/Address : NEST CONSTRUCTION COMPANY LTD Vaitele Uta, Samoa Class(es) 37 Application No. : WS/T/7361 Representation of Mark Filing Date : 24 September, 2018 Priority Date : N/A Proprietor/Address : NIKE INNOVATE C.V. One Bowerman Drive, Beaverton, Oregon 97005--6453, United States of America Class(es) 18 and 25 Application No. : WS/T/7464 Representation of Mark Filing Date : 29 July, 2019 Priority Date : 04/03/2019 Proprietor/Address : REIGN BEVERAGE COMPANY LLC 1547 N. -

Chemical Formula of Binary Ionic Compounds – Sheet 1 the Combining Power Or Valency of Silver Is Always 1
Chemical Formula of Binary Ionic Compounds – Sheet 1 The combining power or valency of silver is always 1. All other transition metals are 2 unless otherwise indicated. No. Binary compound Formula No. Binary compound Formula 1 potassium fluoride 26 calcium sulfide 2 calcium chloride 27 lithium bromide 3 barium bromide 28 nickel sulfide 4 silver sulfide 29 zinc phosphide 5 aluminium iodide 30 barium iodide 6 potassium iodide 31 caesium chloride 7 lead(IV) oxide 32 copper bromide 8 zinc nitride 33 sodium nitride 9 silver iodide 34 silver chloride 10 barium fluoride 35 sodium hydride 11 lead(II) iodide 36 potassium nitride 12 silver fluoride 37 cobalt chloride 13 sodium sulfide 38 magnesium sulfide 14 sodium bromide 39 potassium chloride 15 calcium oxide 40 calcium bromide 16 zinc fluoride 41 iron(III) oxide 17 strontium phosphide 42 aluminium fluoride 18 barium sulfide 43 magnesium bromide 19 aluminium oxide 44 iron(III) chloride 20 aluminium chloride 45 barium nitride 21 aluminium sulfide 46 sodium fluoride 22 lead(II) oxide 47 lithium fluoride 23 barium chloride 48 lithium iodide 24 copper chloride 49 lithium hydride 25 barium phosphide 50 potassium oxide “Aluminum” and “cesium” are commonly used alternative spellings for "aluminium" and "caesium that are used in the US. May be freely copied for educational use. ©www.chemicalformula.org Chemical Formula of Binary Ionic Compounds – Sheet 2 The combining power or valency of silver is always 1. All other transition metals are 2 unless otherwise indicated. No. Binary compound Formula No. -

Ep 1831144 B1
(19) & (11) EP 1 831 144 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07C 37/50 (2006.01) C07C 39/17 (2006.01) 17.08.2011 Bulletin 2011/33 C07C 39/26 (2006.01) (21) Application number: 05816593.7 (86) International application number: PCT/HU2005/000128 (22) Date of filing: 07.12.2005 (87) International publication number: WO 2006/061666 (15.06.2006 Gazette 2006/24) (54) A NEW PROCESS FOR THE PREPARATION OF PHENOLIC HYDROXY-SUBSTITUTED COMPOUNDS NEUES VERFAHREN ZUR HERSTELLUNG PHENOLISCHER HYDROXY SUBSTITUIERTER VERBINDUNGEN PROCEDE D’ELABORATION DE COMPOSES PHENOLIQUES A SUBSTITUTION HYDROXY (84) Designated Contracting States: • BORBÉLY, László AT BE BG CH CY CZ DE DK EE ES FI FR GB GR H-2510 Dorog (HU) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • HAÁSZ, Ferenc SK TR H-2500 Esztergom (HU) Designated Extension States: • JEKKEL, Péter AL BA HR MK YU H-2509 Esztergom-Kertváros (HU) (30) Priority: 08.12.2004 HU 0402530 (74) Representative: HOFFMANN EITLE 11.11.2005 HU 0501044 Patent- und Rechtsanwälte Arabellastraße 4 (43) Date of publication of application: 81925 München (DE) 12.09.2007 Bulletin 2007/37 (56) References cited: (73) Proprietor: Richter Gedeon Nyrt. US-A- 4 339 614 1103 Budapest (HU) • MANABU NODE ET.AL.: "Hard acid and soft (72) Inventors: nucleophile system. 2. Demethylation of methyl • VASS, András ethers of alcohol and phenol with an aluminium H-8200 Veszprém (HU) halide-thiolsystem" J. ORG. CHEM., vol. 45, 1980, • DUDÁS, József pages 4275-4277, XP002379759 cited in the H-8200 Veszprém (HU) application Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -

Chemical Resistance Table
CHEMICAL RESISTANCE TABLE Things to consider when choosing a hose • This chemical resistance table indicates if the inner tube of the hose is resistant to specific materials/chemicals throughout different temperatures. • Some materials/chemicals can change color in contact with the hose. If it´s important to have the color unaffected, we recommend you to contact Hydroscand. • For food products the table only indicates whether the inner tube is resistant to the product. This doesn’t automatically significate that the inner tube is approved for food products. • Abrasion, friction and mechanical influence can increase the chemicals aggression and therefore decrease the durability of the hose. • All values in the table are exclusively for the transport of media. • NB! Materials can change color in contact with other kind of materials. • Outer stress is always an important factor to the durability of the hose. NB! This resistance table should be considered as an indication and not a guarantee. International material codes Rubber Plasic NR Natural rubber P.T.F.E. Polytetraflouro ethylene (Teflon®) SBR Styrene butadiene rubber PP Polypropylene NBR Nitrile rubber UPE Ultra high molekular weight EPDM Ethylene-propylene rubber polyethylene IIR Butyl rubber XLPE Cross linked polyethylene (PEX) CR Chloroprene rubber (Neopren) PU Polyurethane CSM Chlorosulfonated polyethylene PE Polyester platsic (Elastomer) rubber (Hypalon) PA Polyamide (Nylon) PVC Polyvinylchloride How to read the table Fitness grade A Good to excelent B Acceptable for limited use -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Durham E-Theses
Durham E-Theses Acid induced reactions of polyuoroaromatic compounds Thorpe, John Graham How to cite: Thorpe, John Graham (1969) Acid induced reactions of polyuoroaromatic compounds, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/8656/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk UNIVERSITY OF DURHAM A THESIS entitled ACID INDUCED REACTIONS OF POLYFLUOROAROMATIC COMPOUNDS Submitted by JOHN GRAHAM THORPE, B.Sc. (UNIVERSITY COLLEGE) A candidate for the degree of Doctor of Philosophy. 1969. ACKNOWLEDGEMENTS The author would like to express his gratitude to Professor W.K.R. Musgrave and Dr. R.D. Chambers for their continual help and encouragement during the supervision of this work, and to Dr. M. Hole for many helpful discussions and valuable advice. Thanks are also due to many technical and laboratory staff for their help, and to Imperial Smelting Corporation Limited for a maintenance grant. -

Recent Advances in Low Oxidation State Aluminium Chemistry Cite This: Chem
Chemical Science PERSPECTIVE View Article Online View Journal | View Issue Recent advances in low oxidation state aluminium chemistry Cite this: Chem. Sci., 2020, 11, 6942 All publication charges for this article Katie Hobson, Claire J. Carmalt and Clare Bakewell * have been paid for by the Royal Society of Chemistry The synthesis and isolation of novel low oxidation state aluminium (Al) compounds has seen relatively slow progress over the 30 years since such species were first isolated. This is largely due to the significant challenges in isolating these thermodynamically unstable compounds. Despite challenges with isolation, their reactivity has been widely explored and they have been utilized in a wide range of processes including the activation of strong chemicals bonds, as ligands to transition metals and in the formation of Received 11th May 2020 heterobimetallic M–M compounds. As such, attempts to isolate novel low oxidation state Al compounds Accepted 23rd June 2020 have continued in earnest and in the last few years huge advances have been made. In this review we DOI: 10.1039/d0sc02686g highlight the remarkable recent developments in the low oxidation state chemistry of aluminium and rsc.li/chemical-science discuss the variety of new reactions these compounds have made possible. Creative Commons Attribution 3.0 Unported Licence. Introduction species are one such class of main-group compound whose popularity has soared in recent years. As one of the most Recent years have seen a renaissance in main-group chemistry, abundant elements in the Earth's crust, aluminium's low cost with novel main-group compounds being shown to adopt and low toxicity make it an attractive choice for a wide range of unusual electronic congurations, demonstrate application in applications, from construction and electronics, to a key 8,9 the activation of strong chemical bonds and facilitate a wide component in organic synthesis. -

0620 W20 Qp 13.Pdf
Cambridge IGCSE™ CHEMISTRY 0620/13 Paper 1 Multiple Choice (Core) October/November 2020 45 minutes You must answer on the multiple choice answer sheet. *4297468909* You will need: Multiple choice answer sheet Soft clean eraser Soft pencil (type B or HB is recommended) INSTRUCTIONS There are forty questions on this paper. Answer all questions. For each question there are four possible answers A, B, C and D. Choose the one you consider correct and record your choice in soft pencil on the multiple choice answer sheet. Follow the instructions on the multiple choice answer sheet. Write in soft pencil. Write your name, centre number and candidate number on the multiple choice answer sheet in the spaces provided unless this has been done for you. Do not use correction fluid. Do not write on any bar codes. You may use a calculator. INFORMATION The total mark for this paper is 40. Each correct answer will score one mark. A mark will not be deducted for a wrong answer. Any rough working should be done on this question paper. The Periodic Table is printed in the question paper. This document has 16 pages. Blank pages are indicated. IB20 11_0620_13/4RP © UCLES 2020 [Turn over 2 1 ‘The movement of a substance very slowly from an area of high concentration to an area of low concentration.’ Which process is being described? A a liquid being frozen B a solid melting C a substance diffusing through a liquid D a substance diffusing through the air 2 When a dark grey solid element is heated, it changes directly into a purple gas. -
![United States Patent [191 [11] Patent Number: 4,773,973 Griiniger Et Al](https://docslib.b-cdn.net/cover/5401/united-states-patent-191-11-patent-number-4-773-973-griiniger-et-al-5025401.webp)
United States Patent [191 [11] Patent Number: 4,773,973 Griiniger Et Al
United States Patent [191 [11] Patent Number: 4,773,973 Griiniger et al. [45] Date of Patent: Sep. 27, 1988 [54] PROCESS FOR THE PRODUCTION OF POLYCRYSTALLINE SILICON COATINGS FOREIGN PATENT DOCUMENTS BY ELECTROLYTIC DEPOSITION OF 10415 l/l983 European Pat. Off. SILICON OTHER PUBLICATIONS [75] Inventors: Hans R. Griiniger, Olten; Rudolf Monnier, Chimia, 105-124 (1983). Kern, Maggia; Paul Rys, Zurich, all Elswell, J. Crystal Growth, 52, 7l4-752 (1981). of Sweden Dennis Elwell et al., Solar Energy Materials, 6, pp. 123-145, (1982). [73] Assignee: Ciba-Geigy AG, Basel, Switzerland D. Elimarskii et al., Chemical Abstracts, 96: 42968j (1982). [21] Appl. No.: 165,492 Primary Examiner-G. L. Kaplan . [22] ‘ Filed: Mar. 8, 1988 Attorney, Agent, or Firm—Wenderoth, Lind & Ponack [57] ABSTRACT Related US. Application Data A novel process for the electrolytic deposition of silicon [62] Division of Ser. No. 87,635, Aug. 18, 1987. from a melt containing covalent silicon compounds, in particular silicon tetrahalides, and furthermore alumin [30] Foreign Application Priority Data ium halides, alkali metal halides and halides of transition metals is carried out at relatively low temperatures of Aug. 19, 1986 [CH] Switzerland ....................... .. 3320/86 100° to 350° C. in an inert atmosphere. The silicon is deposited cathodically or anodically onto electrically [51] Int. Cl.4 .............................................. .. C25D 3/66 [52] US. Cl. 204/39 conductive material. [58] Field of Search .................................. .. 204/39, 60 The silicon coatings are homogeneous and adhere ?rmly to the substrate. The coated materials can be used [56] References Cited for the production of photoconductive or photovoltaic devices. U.S. -
5070 W20 Qp 21.Pdf
Cambridge O Level *4652328465* CHEMISTRY 5070/21 Paper 2 Theory October/November 2020 1 hour 30 minutes You must answer on the question paper. No additional materials are needed. INSTRUCTIONS ● Section A: answer all questions. ● Section B: answer three questions. ● Use a black or dark blue pen. You may use an HB pencil for any diagrams or graphs. ● Write your name, centre number and candidate number in the boxes at the top of the page. ● Write your answer to each question in the space provided. ● Do not use an erasable pen or correction fluid. ● Do not write on any bar codes. ● You may use a calculator. ● You should show all your working and use appropriate units. INFORMATION ● The total mark for this paper is 75. ● The number of marks for each question or part question is shown in brackets [ ]. ● The Periodic Table is printed in the question paper. This document has 20 pages. Blank pages are indicated. DC (CE/FC) 184044/4 © UCLES 2020 [Turn over 2 Section A Answer all the questions in this section in the spaces provided. The total mark for this section is 45. 1 Choose from the following compounds to answer the questions. aluminium iodide ethanol glucose lead(IV) chloride lithium bromide magnesium carbonate methane potassium phosphate silver nitrate sodium sulfate sulfur dioxide Each compound may be used once, more than once or not at all. Which compound: (a) produces ammonia when its aqueous solution is warmed with aqueous sodium hydroxide and aluminium ............................................................................................................................................. [1] (b) contains ions with a 1– charge which are present in many fertilisers ............................................................................................................................................