The Halogen Chemistry of the Actinides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Alternate Graphical Representation of Periodic Table of Chemical Elements Mohd Abubakr1, Microsoft India (R&D) Pvt
An Alternate Graphical Representation of Periodic table of Chemical Elements Mohd Abubakr1, Microsoft India (R&D) Pvt. Ltd, Hyderabad, India. [email protected] Abstract Periodic table of chemical elements symbolizes an elegant graphical representation of symmetry at atomic level and provides an overview on arrangement of electrons. It started merely as tabular representation of chemical elements, later got strengthened with quantum mechanical description of atomic structure and recent studies have revealed that periodic table can be formulated using SO(4,2) SU(2) group. IUPAC, the governing body in Chemistry, doesn‟t approve any periodic table as a standard periodic table. The only specific recommendation provided by IUPAC is that the periodic table should follow the 1 to 18 group numbering. In this technical paper, we describe a new graphical representation of periodic table, referred as „Circular form of Periodic table‟. The advantages of circular form of periodic table over other representations are discussed along with a brief discussion on history of periodic tables. 1. Introduction The profoundness of inherent symmetry in nature can be seen at different depths of atomic scales. Periodic table symbolizes one such elegant symmetry existing within the atomic structure of chemical elements. This so called „symmetry‟ within the atomic structures has been widely studied from different prospects and over the last hundreds years more than 700 different graphical representations of Periodic tables have emerged [1]. Each graphical representation of chemical elements attempted to portray certain symmetries in form of columns, rows, spirals, dimensions etc. Out of all the graphical representations, the rectangular form of periodic table (also referred as Long form of periodic table or Modern periodic table) has gained wide acceptance. -

Key Official US and IAEA Statements About Iran's Nuclear Programs
Key Official US and IAEA Statements About Iran’s Nuclear Programs Anthony H. Cordesman There is a great deal of speculation about Iran’s nuclear programs that do not list sources or reflect the views of the US intelligence community. It is worth examining what top US intelligence official have said during the last few years, and the details of the IAEA report published in November 2011 – one that clearly reflected official inputs from the US and a number of European intelligence services. U.S. Official Statements on the Iranian Nuclear and Missile Threat The annual unclassified reports to Congress by the Office of the Director of National Intelligence are a key source of US official view. Although the 2010 report by James R. Clapper has already been partly overtaken by the pace of Iran’s rapidly developing program, it still represents the most detailed unclassified estimate of Iran’s capabilities by a senior US official:1 Nuclear We continue to assess Iran is keeping open the option to develop nuclear weapons though we do not know whether Tehran eventually will decide to produce nuclear weapons. Iran continues to develop a range of capabilities that could be applied to producing nuclear weapons, if a decision is made to do so. During the reporting period, Iran continued to expand its nuclear infrastructure and continued uranium enrichment and activities related to its heavy water research reactor, despite multiple United Nations Security Council Resolutions since late 2006 calling for the suspension of those activities. Although Iran made progress in expanding its nuclear infrastructure during 2001, some obstacles slowed progress during this period. -

Depleted Uranium Hexafluoride: Waste Or Resource?
UCRGJC-120397 PREPRINT Depleted Uranium Hexafluoride: Waste or Resource? N. Schwertz J. Zoller R Rosen S. Patton C. Bradley A. Murray This paper was prepared for submittal to the Global ‘95 International Conference on Evaluation of Emerging Nuclear Fuel Cycle Systems Versailles, France September 11-14,1995 July 1995 This isa preprint of apaper intended for publication in a jaurnal orproceedings. Since changes may be made before publication, this preprint is made available with the understanding that it will not be cited or reproduced without the permiasion of the anthor. DISCLAIMER This document was prepared as an account of work sponsored by an agency of the United Stat= Government. Neither theunited States Governmentmor theuniversity of California nor any oftheir employees, makes any warranty, express or implied, or assumesanylegalliabilityorrespomibility forthe accuracy,completeness,orusefuin~ of any information, apparatus, pduct, or process disdosed, or represents that its use wouldnotinfringe privatelyowned rights. Referencehemin to anyspe&c commercial prodocis, proms, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constituteor imply its endorsement, reconunendation, or favoring by the United States Government or the University of California. The views and opinions of authors expressed herein do not necessady state or reflect those of the United States Government or the University of California, and shall not be used for adveltising or product endorsement purposes. DISCLAIMER Portions of this document may be illegible in electronic image products. Images are produced from the best available original document . DEPLETED URANIUM HEXAFLUORIDE: WASTE OR RESOURCE? N. Schwertz, J. Zoller, R. Rosen, S. Patton LAWRENCE LIVERMORE NATIONAL LABORATORY P. -

SAFETY DATA SHEET Revision Date 28.07.2021 According to Regulation (EC) No
Version 7.0 SAFETY DATA SHEET Revision Date 28.07.2021 according to Regulation (EC) No. 1907/2006 Print Date 02.10.2021 GENERIC EU MSDS - NO COUNTRY SPECIFIC DATA - NO OEL DATA SECTION 1: Identification of the substance/mixture and of the company/undertaking 1.1 Product identifiers Product name : Aluminum iodide Product Number : 208493 Brand : Aldrich REACH No. : A registration number is not available for this substance as the substance or its uses are exempted from registration, the annual tonnage does not require a registration or the registration is envisaged for a later registration deadline. CAS-No. : 7784-23-8 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses : Laboratory chemicals, Manufacture of substances 1.3 Details of the supplier of the safety data sheet Company : Sigma-Aldrich Inc. 3050 SPRUCE ST ST. LOUIS MO 63103 UNITED STATES Telephone : +1 314 771-5765 Fax : +1 800 325-5052 1.4 Emergency telephone Emergency Phone # : 800-424-9300 CHEMTREC (USA) +1-703- 527-3887 CHEMTREC (International) 24 Hours/day; 7 Days/week SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 Skin corrosion (Sub-category 1B), H314 Serious eye damage (Category 1), H318 For the full text of the H-Statements mentioned in this Section, see Section 16. 2.2 Label elements Labelling according Regulation (EC) No 1272/2008 Aldrich- 208493 Page 1 of 8 The life science business of Merck operates as MilliporeSigma in the US and Canada Pictogram Signal word Danger Hazard statement(s) H314 Causes severe skin burns and eye damage. -

Durham E-Theses
Durham E-Theses Halogenated diazines and triazines Wood, D. E. How to cite: Wood, D. E. (1978) Halogenated diazines and triazines, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/8324/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk UNIVERSITY OF Du'RKAM A THESIS entitled HALOGENATED DIAZINES AND TRIAZINES Submitted by D E. WOOD (Grey), B Sc (London) The copyright of this thesis rests with the author No quotation from it should be published without his prior written consent and information derived from it should be acknowledged A candidate for the degree of Doctor of Philosophy 19 78 sr i i > j J To my MoLhcr and FaLhcr WLLII Lh.inks for .ill LhaL Lhey have done ACKNOWLEDGEMLNTS I would like LO express my thanks Lo Professor R D Chambers i under whose guidance this research was undertaken, for considerable encouragement, advice and discussion Thanks are due to Dr R S Matthews for his expert advice with n in r. -

1 Abietic Acid R Abrasive Silica for Polishing DR Acenaphthene M (LC
1 abietic acid R abrasive silica for polishing DR acenaphthene M (LC) acenaphthene quinone R acenaphthylene R acetal (see 1,1-diethoxyethane) acetaldehyde M (FC) acetaldehyde-d (CH3CDO) R acetaldehyde dimethyl acetal CH acetaldoxime R acetamide M (LC) acetamidinium chloride R acetamidoacrylic acid 2- NB acetamidobenzaldehyde p- R acetamidobenzenesulfonyl chloride 4- R acetamidodeoxythioglucopyranose triacetate 2- -2- -1- -β-D- 3,4,6- AB acetamidomethylthiazole 2- -4- PB acetanilide M (LC) acetazolamide R acetdimethylamide see dimethylacetamide, N,N- acethydrazide R acetic acid M (solv) acetic anhydride M (FC) acetmethylamide see methylacetamide, N- acetoacetamide R acetoacetanilide R acetoacetic acid, lithium salt R acetobromoglucose -α-D- NB acetohydroxamic acid R acetoin R acetol (hydroxyacetone) R acetonaphthalide (α)R acetone M (solv) acetone ,A.R. M (solv) acetone-d6 RM acetone cyanohydrin R acetonedicarboxylic acid ,dimethyl ester R acetonedicarboxylic acid -1,3- R acetone dimethyl acetal see dimethoxypropane 2,2- acetonitrile M (solv) acetonitrile-d3 RM acetonylacetone see hexanedione 2,5- acetonylbenzylhydroxycoumarin (3-(α- -4- R acetophenone M (LC) acetophenone oxime R acetophenone trimethylsilyl enol ether see phenyltrimethylsilyl... acetoxyacetone (oxopropyl acetate 2-) R acetoxybenzoic acid 4- DS acetoxynaphthoic acid 6- -2- R 2 acetylacetaldehyde dimethylacetal R acetylacetone (pentanedione -2,4-) M (C) acetylbenzonitrile p- R acetylbiphenyl 4- see phenylacetophenone, p- acetyl bromide M (FC) acetylbromothiophene 2- -5- -

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -
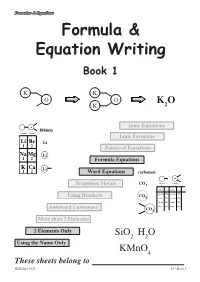
Formula & Equation Writing
Formulae & Equations Formula & Equation Writing Book 1 K K O O K O K 2 Ionic Equations lithium column column 1 2 Ionic Formulae Li Be Li 1 2 Balanced Equations Na Mg Li 1 2 Formula Equations K Ca Li 1 2 Word Equations carbonate valency valency Transition Metals CO3 1 2 name formula name formula ammonium NH4 carbonate CO3 Using Brackets CO 3 cyanide CN chromate CrO4 hydroxide OH sulphate SO4 nitrate NO sulphite SO Awkward Customers 3 3 CO3 More than 2 Elements 2 Elements Only SiO2 H2O Using the Name Only KMnO4 These sheets belong to KHS Sept 2013 page 1 S3 - Book 1 Formulae & Equations What is a Formula ? The formula of a compound tells you two things about the compound :- SiO H O i) which elements are in the compound 2 2 using symbols, and KMnO4 ii) how many atoms of each element are in the compound using numbers. Test Yourself What would be the formula for each of the following? Br H Cl K H C S Al Br O H Cl K H Br Using the Name Only Some compounds have extra information in their carbon monoxide names that allow people to work out and write CO the correct formula. carbon dioxide CO2 The names of the elements appear as usual but dinitrogen tetroxide N O this time the number of each type of atom is 2 4 included using mono- = 1 di- = 12 tri- = 3 tetra- = 4 penta- = 5 hexa- =46 Test Yourself 1 What would be the formula for each of the following? 1. -
NOI to Prepare an Environmental Impact Statement For
Federal Register / Vol. 64, No. 34 / Monday, February 22, 1999 / Notices 8553 reauthorization of OERI and will hear DEPARTMENT OF ENERGY Orders are summarized in the attached committee and officers' reports. A final appendix. [FE Docket Nos. 99±01±NG, 99±03±NG, 99± agenda will be available from the Board 02±NG, 99±04±NG, 92±24±NG, 99±05±NG, These Orders may be found on the FE office on March 10, 1999, and will be and 99±06±NG] web site at http://www.fe.doe.gov., or posted on the Board's web site, http:// on the electronic bulletin board at (202) www.ed.gov/offices/OERI/NERPPB/. Office of Fossil Energy; Orders 586±7853. Granting and Transferring Records are kept of all Board They are also available for inspection Authorizations To Import and/or Export proceedings and are available for public and copying in the Office of Natural Gas Natural Gas inspection at the office of the National & Petroleum Import & Export Activities, Educational Research Policy and OGE Energy Resources, Inc., National Fuel Docket Room 3E±033, Forrestal Priorities Board, Suite 100, 80 F St., Gas Distribution Corporation, Renaissance Building, 1000 Independence Avenue, NW, Washington, D.C. 20208±7564. Energy (U.S.) Inc., Selkirk Cogen Partners, SW, Washington, DC 20585, (202) 586± L.P., Coral Energy Resources, L.P. (Successor 9478. The Docket Room is open between Dated: February 16, 1999. to Salmon Resources Ltd.), Transco Energy the hours of 8:00 a.m. and 4:30 p.m., Eve M. Bither, Marketing Company, and Petro-Canada Hydrocarbons Inc. -

The Oxidizing Behavior of Some Platinum Metal Fluorides
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title THE OXIDIZING BEHAVIOR OF SOME PLATINUM METAL FLUORIDES Permalink https://escholarship.org/uc/item/46b5r4v5 Author Graham, Lionell Publication Date 1978-10-01 eScholarship.org Powered by the California Digital Library University of California LBL-8088.c,y '': ·,~, . ' '~~.. .· J!' ... THE OXIDIZING BEHAV!OROF SOME PLATINUM METAL FLUORIDES . Li onell Graham. · (Ph~ D. Thesis ) IIi !l1/ "~ · r-,> ·1cr7o October 1978 ~t .. ~, ~L l ... ;,() · .. t .. J r.~~ r: !\ ~;~, ·l .• ~~:J"~~ t~J ;,~.)(·_ .. {·~ tJ 1v~ ;;:: r~- J··r·r,;~ Ei:~-:·~,:~~ <'11 ().i\{ Prep~red for the .. U. S. Department of Energy. under Contract W~7405~ENG-48 TWO-WEEK LOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. .. ' ·t•·! •. ·. For a personal retention copy, call Tech. Info. Diu is ion, Ext. 6782 .. LEGAL NOTICE -___,..-----~__, This report was prepared as an account of work sponsored by the United States Government Neither the United States nor the Depart ment of Energy, nor any of their employees, nor any of their con tractors, subcontractors, or their employees, makes any warraf')ty, express or implied,orassumes any legal liabii bility for the accuracy, completeness or usefulness of nformation, appa- ratus, product. ·· · · disclosed; or rep ·. ts that its use would. not infringe p. · · owned rights ... -i- THE OXIDIZING BEHAVIOR OF SOME PLATINUM METAL FLUORIDES Contents Abstract . v I. General Introduction 1 II. General Apparatus and Handling Techniques 2 A. Apparatus . 2 1. General 2 2. The Cryogenic Technology, Inc., Model 21 Cryo-Cooler .. 3 3. Reactor for Gas-Gas Reaction Products 5 B. -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Chemistry of the Noble Gases*
CHEMISTRY OF THE NOBLE GASES* By Professor K. K. GREE~woon , :.\I.Sc., sc.D .. r".lU.C. University of N ewca.stle 1tpon Tyne The inert gases, or noble gases as they are elements were unsuccessful, and for over now more appropriately called, are a remark 60 years they epitomized chemical inertness. able group of elements. The lightest, helium, Indeed, their electron configuration, s2p6, was recognized in the gases of the sun before became known as 'the stable octet,' and this it was isolated on ea.rth as its name (i]A.tos) fotmed the basis of the fit·st electronic theory implies. The first inert gas was isolated in of valency in 1916. Despite this, many 1895 by Ramsay and Rayleigh; it was named people felt that it should be possible to induce argon (apy6s, inert) and occurs to the extent the inert gases to form compounds, and many of 0·93% in the earth's atmosphere. The of the early experiments directed to this end other gases were all isolated before the turn have recently been reviewed.l of the century and were named neon (v€ov, There were several reasons why chemists new), krypton (KpVn'TOV, hidden), xenon believed that the inert gases might form ~€vov, stmnger) and radon (radioactive chemical compounds under the correct con emanation). Though they occur much less ditions. For example, the ionization poten abundantly than argon they cannot strictly tial of xenon is actually lower than those of be called rare gases; this can be illustrated hydrogen, nitrogen, oxygen, fl uorine and by calculating the volumes occupied a.t s.t.p.