Population-Level Expression Variability of Mitochondrial DNA-Encoded Genes in Humans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Whole Exome Sequencing in ADHD Trios from Single and Multi-Incident Families Implicates New Candidate Genes and Highlights Polygenic Transmission
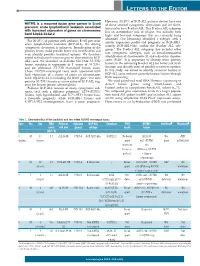
European Journal of Human Genetics (2020) 28:1098–1110 https://doi.org/10.1038/s41431-020-0619-7 ARTICLE Whole exome sequencing in ADHD trios from single and multi-incident families implicates new candidate genes and highlights polygenic transmission 1,2 1 1,2 1,3 1 Bashayer R. Al-Mubarak ● Aisha Omar ● Batoul Baz ● Basma Al-Abdulaziz ● Amna I. Magrashi ● 1,2 2 2,4 2,4,5 6 Eman Al-Yemni ● Amjad Jabaan ● Dorota Monies ● Mohamed Abouelhoda ● Dejene Abebe ● 7 1,2 Mohammad Ghaziuddin ● Nada A. Al-Tassan Received: 26 September 2019 / Revised: 26 February 2020 / Accepted: 10 March 2020 / Published online: 1 April 2020 © The Author(s) 2020. This article is published with open access Abstract Several types of genetic alterations occurring at numerous loci have been described in attention deficit hyperactivity disorder (ADHD). However, the role of rare single nucleotide variants (SNVs) remains under investigated. Here, we sought to identify rare SNVs with predicted deleterious effect that may contribute to ADHD risk. We chose to study ADHD families (including multi-incident) from a population with a high rate of consanguinity in which genetic risk factors tend to 1234567890();,: 1234567890();,: accumulate and therefore increasing the chance of detecting risk alleles. We employed whole exome sequencing (WES) to interrogate the entire coding region of 16 trios with ADHD. We also performed enrichment analysis on our final list of genes to identify the overrepresented biological processes. A total of 32 rare variants with predicted damaging effect were identified in 31 genes. At least two variants were detected per proband, most of which were not exclusive to the affected individuals. -
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

Viewed and Published Immediately Upon Acceptance Cited in Pubmed and Archived on Pubmed Central Yours — You Keep the Copyright
BMC Genomics BioMed Central Research article Open Access Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice Claudia Prinzen1, Dietrich Trümbach2, Wolfgang Wurst2, Kristina Endres1, Rolf Postina1 and Falk Fahrenholz*1 Address: 1Johannes Gutenberg-University, Institute of Biochemistry, Mainz, Johann-Joachim-Becherweg 30, 55128 Mainz, Germany and 2Helmholtz Zentrum München – German Research Center for Environmental Health, Institute for Developmental Genetics, Ingolstädter Landstraße 1, 85764 Neuherberg, Germany Email: Claudia Prinzen - [email protected]; Dietrich Trümbach - [email protected]; Wolfgang Wurst - [email protected]; Kristina Endres - [email protected]; Rolf Postina - [email protected]; Falk Fahrenholz* - [email protected] * Corresponding author Published: 5 February 2009 Received: 19 June 2008 Accepted: 5 February 2009 BMC Genomics 2009, 10:66 doi:10.1186/1471-2164-10-66 This article is available from: http://www.biomedcentral.com/1471-2164/10/66 © 2009 Prinzen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In a transgenic mouse model of Alzheimer disease (AD), cleavage of the amyloid precursor protein (APP) by the α-secretase ADAM10 prevented amyloid plaque formation, and alleviated cognitive deficits. Furthermore, ADAM10 overexpression increased the cortical synaptogenesis. These results suggest that upregulation of ADAM10 in the brain has beneficial effects on AD pathology. Results: To assess the influence of ADAM10 on the gene expression profile in the brain, we performed a microarray analysis using RNA isolated from brains of five months old mice overexpressing either the α-secretase ADAM10, or a dominant-negative mutant (dn) of this enzyme. -

Molecular Mechanisms in Muscular Dystrophy : a Gene Expression Profiling Study Turk, R
Molecular mechanisms in muscular dystrophy : a gene expression profiling study Turk, R. Citation Turk, R. (2006, September 27). Molecular mechanisms in muscular dystrophy : a gene expression profiling study. Retrieved from https://hdl.handle.net/1887/4577 Version: Corrected Publisher’s Version Licence agreement concerning inclusion of doctoral License: thesis in the Institutional Repository of the University of Leiden Downloaded from: https://hdl.handle.net/1887/4577 Note: To cite this publication please use the final published version (if applicable). Molecular Mechanisms In Muscular Dystrophy A Gene Expression Profiling Study Molecular Mechanisms In Muscular Dystrophy A Gene Expression Profiling Study Proefschrift ter verkrijging van de graad van Doctor aan de Universiteit Leiden, op gezag van de Rector Magnificus Dr. D.D.Breimer, hoogleraar in de faculteit der Wiskunde en Natuurwetenschappen en die der Geneeskunde, volgens besluit van het College voor Promoties te verdedigen op woensdag 27 september 2006 klokke 15.00 uur door Rolf Turk Geboren te Leiden in 1975 Promotiecommissie Promotor Prof. Dr. G.J.B. van Ommen Co-promotores Dr. J.T. den Dunnen Dr. P.A.C. ‘t Hoen Referent Prof. Dr. R.M.W. Hofstra (Rijksuniversiteit Groningen) Overige leden Prof. Dr. M. Koenig (Université Louis Pasteur de Strasbourg) An experiment is a question which science poses to Nature, and a measurement is the recording of Nature’s answer. Max Planck Aan Maaike, Gerard en Annet Printed by: Drukkerij Duineveld ISBN-10: 90-9021042-3 ISBN-13: 978-90-9021042-1 Turk, Rolf Molecular mechanisms in muscular dystrophy. A gene expression profiling study. Thesis, Leiden University Medical Center September 27, 2006 © Rolf Turk No part of this thesis may be reproduced or transmitted in any form or by any means, without the written permission of the copyright owner Molecular Mechanisms In Muscular Dystrophies Preface 9 Chapter 1 Introduction 11 1. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Rabbit Anti-API5/FITC Conjugated Antibody-SL1252R-FITC
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-API5/FITC Conjugated antibody SL1252R-FITC Product Name: Anti-API5/FITC Chinese Name: FITC标记的凋亡抑制因子5抗体 apoptosis inhibitor 5; apoptosis inhibitory protein 5; API5; AAC-11; AAC11; API5L1; AAC 11; Antiapoptosis clone 11 protein; API5 like 1; Cell migration inducing gene 8 Alias: protein; Fibroblast growth factor 2 interacting factor 2; FIF; Migration inducing protein MIG8; MIG 8; MIG8; Protein XAGL; API5_HUMAN. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Pig,Cow,Horse,Guinea Pig, IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 57kDa Form: Lyophilized or Liquid111 Concentration: 1mg/ml immunogen: KLH conjugated synthetic peptide derived from human API5 Lsotype: IgG Purification: affinitywww.sunlongbiotech.com purified by Protein A Storage Buffer: 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: Apoptosis inhibitor 5 (API5) is an antiapoptotic factor which may have a role in protein assembly. API5 expression is often upregulated in tumor cells, particularly in metastatic Product Detail: cells. It is a critical determinant of E2F1-induced apoptosis, acting downstream of E2F to suppress E2F-dependent apoptosis without generally blocking E2F-dependent transcription. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy
International Journal of Molecular Sciences Article A Transcriptome-Wide Isoform Landscape of Melanocytic Nevi and Primary Melanomas Identifies Gene Isoforms Associated with Malignancy Siras Hakobyan 1 , Henry Loeffler-Wirth 2, Arsen Arakelyan 1 , Hans Binder 2,† and Manfred Kunz 3,*,† 1 Institute of Molecular Biology NAS RA, Yerevan 0014, Armenia; [email protected] (S.H.); [email protected] (A.A.) 2 Interdisciplinary Centre for Bioinformatics, University of Leipzig, Härtelstr. 16–18, 04107 Leipzig, Germany; [email protected] (H.L.-W.); [email protected] (H.B.) 3 Department of Dermatology, Venereology and Allergology, University of Leipzig Medical Center, Philipp-Rosenthal-Str. 23, 04103 Leipzig, Germany * Correspondence: [email protected]; Tel.: +49-341-9718610; Fax: +49-341-9718609 † Shared senior authorship. Abstract: Genetic splice variants have become of central interest in recent years, as they play an important role in different cancers. Little is known about splice variants in melanoma. Here, we analyzed a genome-wide transcriptomic dataset of benign melanocytic nevi and primary melanomas (n = 80) for the expression of specific splice variants. Using kallisto, a map for differentially expressed splice variants in melanoma vs. benign melanocytic nevi was generated. Among the top genes with differentially expressed splice variants were Ras-related in brain 6B (RAB6B), a member of Citation: Hakobyan, S.; the RAS family of GTPases, Macrophage Scavenger Receptor 1 (MSR1), Collagen Type XI Alpha 2 Loeffler-Wirth, H.; Arakelyan, A.; Chain (COLL11A2), and LY6/PLAUR Domain Containing 1 (LYPD1). The Gene Ontology terms of Binder, H.; Kunz, M. A differentially expressed splice variants showed no enrichment for functional gene sets of melanoma Transcriptome-Wide Isoform vs. -

UC Santa Cruz UC Santa Cruz Electronic Theses and Dissertations
UC Santa Cruz UC Santa Cruz Electronic Theses and Dissertations Title Global Analysis of U2AF2-RNA Interactions Permalink https://escholarship.org/uc/item/7z01m3x3 Author Kim, Garam Publication Date 2018 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA SANTA CRUZ GLOBAL ANALYSIS OF U2AF2-RNA INTERACTIONS A thesis submitted in partial satisfaction of the requirements for the degree of MASTER OF ARTS in MOLECULAR, CELLULAR, DEVELOPMENTAL BIOLOGY by Garam Kim June 2018 The Thesis of Garam Kim is approved: _______________________________ Professor Jeremy Sanford _______________________________ Professor Grant Hartzog _______________________________ Professor Alan Zahler _______________________________ Tryus Miller Vice Provost and Dean of Graduate Studies Copyright © By Garam Kim 2018 Table of Contents List of Figures v List of Tables vi Abstract vii Dedication viii Acknowledgments ix 1 Introduction 1 1.1 Eukaryotic gene expression. 1 1.2 Pre-mRNA Splicing. 2 1.3 Alternative Splicing. 4 1.4 Role of Alu elements in Primate Evolution. 6 1.5 Comparative Transcriptomics. 8 2 Materials and Methods 11 2.1 Cell Lines and Cell Culture. 11 2.2 Cell Extract Preparation. 12 2.3 Western Blot. 12 2.4 RNA Isolation and RT-PCR. 13 2.5 Validation of hnRNP A1 Regulated Splicing. 14 2.5.1 Primer Design. 14 2.6 β-globin Reporter Assay. 16 2.6.1 Reporter Design. 16 2.7 Individual-Nucleotide Resolution Crosslinking and Immunoprecipitation. 17 2.7.1 Preparation of Crosslinked Cell Extracts. 17 2.7.2 Bead Preparation and Immunoprecipitation. 18 2.7.3 Phenol-Chloroform Extraction of Size Matched Inputs. -

The Neurodegenerative Diseases ALS and SMA Are Linked at The
Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gky1093 The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gky1093/5162471 by [email protected] on 06 November 2018 Binkai Chi, Jeremy D. O’Connell, Alexander D. Iocolano, Jordan A. Coady, Yong Yu, Jaya Gangopadhyay, Steven P. Gygi and Robin Reed* Department of Cell Biology, Harvard Medical School, 240 Longwood Ave. Boston MA 02115, USA Received July 17, 2018; Revised October 16, 2018; Editorial Decision October 18, 2018; Accepted October 19, 2018 ABSTRACT Fused in Sarcoma (FUS) and TAR DNA Binding Protein (TARDBP) (9–13). FUS is one of the three members of Understanding the molecular pathways disrupted in the structurally related FET (FUS, EWSR1 and TAF15) motor neuron diseases is urgently needed. Here, we family of RNA/DNA binding proteins (14). In addition to employed CRISPR knockout (KO) to investigate the the RNA/DNA binding domains, the FET proteins also functions of four ALS-causative RNA/DNA binding contain low-complexity domains, and these domains are proteins (FUS, EWSR1, TAF15 and MATR3) within the thought to be involved in ALS pathogenesis (5,15). In light RNAP II/U1 snRNP machinery. We found that each of of the discovery that mutations in FUS are ALS-causative, these structurally related proteins has distinct roles several groups carried out studies to determine whether the with FUS KO resulting in loss of U1 snRNP and the other two members of the FET family, TATA-Box Bind- SMN complex, EWSR1 KO causing dissociation of ing Protein Associated Factor 15 (TAF15) and EWS RNA the tRNA ligase complex, and TAF15 KO resulting in Binding Protein 1 (EWSR1), have a role in ALS. -

Bioinformatics-Based Prediction of FUT8 As a Therapeutic Target in Estrogen Receptor-Positive Breast Cancer Fateme Shaabanpour Aghamaleki1, Shirin Farivar1,*
Multidisciplinary Cancer Investigation Original Article January 2019, Volume 3, Issue 1 Bioinformatics-Based Prediction of FUT8 as a Therapeutic Target in Estrogen Receptor-Positive Breast Cancer Fateme Shaabanpour Aghamaleki1, Shirin Farivar1,* 1 Department of Cellullar-Molecular Biology , Faculty of biological Sciences and technologies, Shahid Beheshti University G.C., Tehran, Iran *Corresponding author: Shirin Farivar,Department of Cellullar-Molecular Biology, Faculty of biological Sciences and technologies, Shahid Beheshti University G.C., Tehran, Iran. Tel: +982129902720; Fax: +982122431664; E-mail: [email protected] DOI: 10.30699/acadpub.mci.3.1.25 Abstract Submitted: 20 October 2018 Introduction: Estrogen receptor-positive (ER-positive) breast cancer is a subgroup of Revised: 4 November 2018 breast tumors that is more likely to respond to hormone therapy. ER-positive and ER- Accepted: 28 November 2018 negative breast cancers tend to show different patterns of metastasis because of different e-Published: 1 January 2019 signaling cascade and genes that are activated by estrogen response. Genetic factors can contribute to high rates of metastasis in ER-positive breast cancer. Fucosyltransferase 8 Keywords: (FUT8) is a member of fucosyltransferases family and plays an important role in α-1,6 Fucosyltransferases linkage to the first GlcNAc residue of N-glycans chain. In this study, for the first time, Breast Neoplasms Microarray Analysis we predicted FUT8 by bioinformatics tools as a novel therapeutic target for ER-positive Signal Transduction breast cancer. Methods: Microarray gene expression data of 9 patients with ER+ve and 10 individuals with ER-ve breast cancer was extracted from Geodatasets. Gene expression of two ER+ and ER- patients was compared with logfc and then sorted by their p-values. -

HNRNPA1 Promotes Recognition of Splice Site Decoys by U2AF2 In
bioRxiv preprint doi: https://doi.org/10.1101/175901; this version posted August 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 HNRNPA1 promotes recognition of splice site decoys by U2AF2 in 2 vivo 3 Jonathan M. Howard1*, Hai Lin2*, Garam Kim1, Jolene M Draper1, Maximilian Haeussler, Sol 4 Katzman3, Masoud Toloue4, Yunlong Liu2 and Jeremy R. Sanford1 5 Affiliations: 6 1. Department of Molecular, Cellular and Developmental Biology, University of California Santa 7 Cruz, 1156 High Street, Santa Cruz CA 95064 8 2. Department of Medical and Molecular Genetics, Indiana University School of Medicine, 9 Indianapolis, IN 46202, USA 10 3. Center for Biomolecular Science and Engineering, UC Santa Cruz 1156 High Street, Santa 11 Cruz CA 95064 12 4. Bioo Scientific Corporation, 7500 Burleston Rd, Austin, TX, 78744 13 Correspondence to: [email protected] 14 *These authors contributed equally. 15 Email addresses of all authors: [email protected], [email protected], [email protected], 16 [email protected], [email protected], [email protected], [email protected] 17 18 19 20 bioRxiv preprint doi: https://doi.org/10.1101/175901; this version posted August 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license.