Whole-Exome Sequencing Identifies MST1R As a Genetic Susceptibility Gene in Nasopharyngeal Carcinoma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ZMYND10 Functions in a Chaperone Relay During Axonemal Dynein Assembly
bioRxiv preprint doi: https://doi.org/10.1101/233718; this version posted December 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 ZMYND10 functions in a chaperone relay during axonemal dynein assembly. 2 3 Girish R Mali1,9 , Patricia Yeyati1, Seiya Mizuno2, Margaret A Keighren1, Petra zur Lage3, Amaya 4 Garcia-Munoz4, Atsuko Shimada5, Hiroyuki Takeda5, Frank Edlich6, Satoru Takahashi2,7, Alex von 5 Kreigsheim4,8, Andrew Jarman3 and Pleasantine Mill1,*. 6 7 1. MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of 8 Edinburgh, Edinburgh, UK, EH4 2XU 9 2. Laboratory Animal Resource Centre, University of Tsukuba, Tsukuba, Japan, 305-8575 10 3. Centre for Integrative Physiology, University of Edinburgh, Edinburgh, UK, EH8 9XD 11 4. Systems Biology Ireland, University College Dublin, Dublin, Ireland 12 5. Department of Biological Sciences, University of Tokyo, Tokyo, Japan, 113-0033 13 6. Institute for Biochemistry and Molecular Biology, University of Freiburg, Freiburg, Germany, 14 79104 15 7. Department of Anatomy and Embryology, Faculty of Medicine, University of Tsukuba, Tsukuba, 16 Japan, 305-8575 17 8. Edinburgh Cancer Research UK Centre, Institute of Genetics and Molecular Medicine, University 18 of Edinburgh, Edinburgh, UK, EH4 2XU 19 9. Current address: MRC Laboratory of Molecular Biology, Cambridge, UK, CB2 0QH 20 * Corresponding author: [email protected] 21 22 23 24 25 26 27 28 29 30 31 1 bioRxiv preprint doi: https://doi.org/10.1101/233718; this version posted December 13, 2017. -

HOOK3 Is a Scaffold for the Opposite-Polarity Microtubule-Based
bioRxiv preprint doi: https://doi.org/10.1101/508887; this version posted December 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. HOOK3 is a scaffold for the opposite-polarity microtubule-based motors cytoplasmic dynein and KIF1C Agnieszka A. Kendrick1, William B. Redwine1,2†, Phuoc Tien Tran1‡, Laura Pontano Vaites2, Monika Dzieciatkowska4, J. Wade Harper2, and Samara L. Reck-Peterson1,3,5 1Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, CA, 92093. 2 Department of Cell Biology, Harvard Medical School, Boston, MA 02115. 3Section of Cell and Developmental Biology, Division of Biological Sciences, University of California San Diego, La Jolla, CA 92093. 4Department of Biochemistry and Molecular Genetics, University of Colorado Denver, Aurora, CO 80045. 5Howard Hughes Medical Institute †Present address: Stowers Institute for Medical Research, Kansas City, MO 64110 ‡Present address: Department of Molecular and Cellular Biology, Harvard University, Cambridge, MA 02138. *Correspondence to: Samara Reck-Peterson 9500 Gilman Drive, Leichtag 482 La Jolla CA, 92093 [email protected]; https://orcid.org/0000-0002-1553-465X 1 bioRxiv preprint doi: https://doi.org/10.1101/508887; this version posted December 31, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Product Datasheet KIF1C Antibody NB100-57510
Product Datasheet KIF1C Antibody NB100-57510 Unit Size: 0.1 ml Store at 4C. Do not freeze. Reviews: 1 Publications: 1 Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NB100-57510 Updated 3/16/2021 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NB100-57510 Page 1 of 3 v.20.1 Updated 3/16/2021 NB100-57510 KIF1C Antibody Product Information Unit Size 0.1 ml Concentration 0.2 mg/ml Storage Store at 4C. Do not freeze. Clonality Polyclonal Preservative 0.09% Sodium Azide Isotype IgG Purity Immunogen affinity purified Buffer TBS and 0.1% BSA Product Description Host Rabbit Gene ID 10749 Gene Symbol KIF1C Species Human, Mouse Immunogen The immunogen recognized by this antibody maps to a region between residue 1053 and the C-terminus (residue 1103) of human kinesin family member 1C (lethal toxin sensitivity 1) using the numbering given in entry NP_006603.2 (GeneID 10749). Product Application Details Applications Western Blot, Immunoprecipitation Recommended Dilutions Western Blot 1:2000-1:10000, Immunoprecipitation 2 - 5 ug/mg lysate Page 2 of 3 v.20.1 Updated 3/16/2021 Images Western Blot: KIF1C Antibody [NB100-57510] - KIF1C is a novel HOOK3 -interacting protein.sfGFP-3xFLAG and full length (FL) HOOK3, HOOK3 (1-552), and HOOK3 (553-718) all tagged with sfGFP and 3xFLAG were immunoprecipitated with alpha-FLAG antibodies from transiently transfected HEK-293T cells. Western blots with alpha-HC, alpha- FAM160A2, alpha-KIF1C, and alpha-FLAG antibodies were used to determine which proteins co-immunoprecipitated with each HOOK3 construct.DOI:http://dx.doi.org/10.7554/eLife.28257.015 Image collected and cropped by CiteAb from the following publication (https://elifesciences.org/articles/28257), licensed under a CC-BY licence. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Whole Genome Sequencing Identifies Novel Structural Variant in a Large Indian Family Affected with X - Linked Agammaglobulinemia
medRxiv preprint doi: https://doi.org/10.1101/2020.10.05.20200949; this version posted October 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-ND 4.0 International license . Whole Genome Sequencing identifies novel structural variant in a large Indian family affected with X - linked agammaglobulinemia 1,2,# 3,5,#,$ 3,4 3 Abhinav Jain , Geeta Madathil Govindaraj , Athulya Edavazhippurath , Nabeel Faisal , Rahul C 1 1,2 6 3 1 1 Bhoyar , Vishu Gupta , Ramya Uppuluri , Shiny Padinjare Manakkad , Atul Kashyap , Anoop Kumar , 1,2 1,2 7 7 3 Mohit Kumar Divakar , Mohamed Imran , Sneha Sawant , Aparna Dalvi , Krishnan Chakyar , 7 6 1,2,$ 1,2,$ Manisha Madkaikar , Revathi Raj , Sridhar Sivasubbu , Vinod Scaria 1 CSIR-Institute of Genomics and Integrative Biology, Mathura Road, New Delhi, Delhi, India 2 Academy of Scientific and Innovative Research (AcSIR), Mathura Road, New Delhi, Delhi, India 3 Department of Pediatrics, Government Medical College Kozhikode, Kozhikode, Kerala, India 4 Multidisciplinary Research Unit, Government College Kozhikode, Kozhikode, Kerala, India 5 FPID Regional Diagnostic Centre, Government Medical College Kozhikode, Kozhikode, Kerala India 6 Department of Pediatric Hematology, Oncology, Blood and Marrow Transplantation, Apollo Hospitals, 320, Padma complex, Anna salai, Teynampet, Chennai, Tamil Nadu, India 7 Department of Pediatric Immunology and Leukocyte Biology, ICMR-National Institute of Immunohaematology, KEM Hospital, Parel, Mumbai, Maharashtra, India. # Joint first authors $ Corresponding author Vinod Scaria: [email protected] Sridhar Sivasubbu: [email protected] Geeta Madathil Govindaraj: [email protected] NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. -

Whole Exome Sequencing in ADHD Trios from Single and Multi-Incident Families Implicates New Candidate Genes and Highlights Polygenic Transmission
European Journal of Human Genetics (2020) 28:1098–1110 https://doi.org/10.1038/s41431-020-0619-7 ARTICLE Whole exome sequencing in ADHD trios from single and multi-incident families implicates new candidate genes and highlights polygenic transmission 1,2 1 1,2 1,3 1 Bashayer R. Al-Mubarak ● Aisha Omar ● Batoul Baz ● Basma Al-Abdulaziz ● Amna I. Magrashi ● 1,2 2 2,4 2,4,5 6 Eman Al-Yemni ● Amjad Jabaan ● Dorota Monies ● Mohamed Abouelhoda ● Dejene Abebe ● 7 1,2 Mohammad Ghaziuddin ● Nada A. Al-Tassan Received: 26 September 2019 / Revised: 26 February 2020 / Accepted: 10 March 2020 / Published online: 1 April 2020 © The Author(s) 2020. This article is published with open access Abstract Several types of genetic alterations occurring at numerous loci have been described in attention deficit hyperactivity disorder (ADHD). However, the role of rare single nucleotide variants (SNVs) remains under investigated. Here, we sought to identify rare SNVs with predicted deleterious effect that may contribute to ADHD risk. We chose to study ADHD families (including multi-incident) from a population with a high rate of consanguinity in which genetic risk factors tend to 1234567890();,: 1234567890();,: accumulate and therefore increasing the chance of detecting risk alleles. We employed whole exome sequencing (WES) to interrogate the entire coding region of 16 trios with ADHD. We also performed enrichment analysis on our final list of genes to identify the overrepresented biological processes. A total of 32 rare variants with predicted damaging effect were identified in 31 genes. At least two variants were detected per proband, most of which were not exclusive to the affected individuals. -

Exome Sequencing and Functional Validation in Zebrafish Identify
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Exome Sequencing and Functional Validation in Zebrafish Identify GTDC2 Mutations as a Cause of Walker-Warburg Syndrome M. Chiara Manzini,1,2 Dimira E. Tambunan,1,2 R. Sean Hill,1,2 Tim W. Yu,1,2 Thomas M. Maynard,3 Erin L. Heinzen,4 Kevin V. Shianna,4 Christine R. Stevens,5 Jennifer N. Partlow,1,2 Brenda J. Barry,1,2 Jacqueline Rodriguez,1,2 Vandana A. Gupta,1,6 Abdel-Karim Al-Qudah,7 Wafaa M. Eyaid,8 Jan M. Friedman,9,10 Mustafa A. Salih,11 Robin Clark,12 Isabella Moroni,13 Marina Mora,14 Alan H. Beggs,1,6 Stacey B. Gabriel,5 and Christopher A. Walsh1,2,5,* Whole-exome sequencing (WES), which analyzes the coding sequence of most annotated genes in the human genome, is an ideal approach to studying fully penetrant autosomal-recessive diseases, and it has been very powerful in identifying disease-causing muta- tions even when enrollment of affected individuals is limited by reduced survival. In this study, we combined WES with homozygosity analysis of consanguineous pedigrees, which are informative even when a single affected individual is available, to identify genetic mutations responsible for Walker-Warburg syndrome (WWS), a genetically heterogeneous autosomal-recessive disorder that severely affects the development of the brain, eyes, and muscle. Mutations in seven genes are known to cause WWS and explain 50%–60% of cases, but multiple additional genes are expected to be mutated because unexplained cases show suggestive linkage to diverse loci. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Genomic Approaches for Understanding the Genetics of Complex Disease
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Perspective Genomic approaches for understanding the genetics of complex disease William L. Lowe Jr.1 and Timothy E. Reddy2,3 1Division of Endocrinology, Metabolism and Molecular Medicine, Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, Illinois 60611, USA; 2Department of Biostatistics and Bioinformatics, Duke University Medical School, Durham, North Carolina 27708, USA; 3Center for Genomic and Computational Biology, Duke University Medical School, Durham, North Carolina 27708, USA There are thousands of known associations between genetic variants and complex human phenotypes, and the rate of novel discoveries is rapidly increasing. Translating those associations into knowledge of disease mechanisms remains a fundamental challenge because the associated variants are overwhelmingly in noncoding regions of the genome where we have few guiding principles to predict their function. Intersecting the compendium of identified genetic associations with maps of regulatory activity across the human genome has revealed that phenotype-associated variants are highly enriched in candidate regula- tory elements. Allele-specific analyses of gene regulation can further prioritize variants that likely have a functional effect on disease mechanisms; and emerging high-throughput assays to quantify the activity of candidate regulatory elements are a promising next step in that direction. Together, these technologies have created the ability to systematically and empirically test hypotheses about the function of noncoding variants and haplotypes at the scale needed for comprehensive and system- atic follow-up of genetic association studies. Major coordinated efforts to quantify regulatory mechanisms across genetically diverse populations in increasingly realistic cell models would be highly beneficial to realize that potential. -

Supplementary Information Method CLEAR-CLIP. Mouse Keratinocytes
Supplementary Information Method CLEAR-CLIP. Mouse keratinocytes of the designated genotype were maintained in E-low calcium medium. Inducible cells were treated with 3 ug/ml final concentration doxycycline for 24 hours before performing CLEAR-CLIP. One 15cm dish of confluent cells was used per sample. Cells were washed once with cold PBS. 10mls of cold PBS was then added and cells were irradiated with 300mJ/cm2 UVC (254nM wavelength). Cells were then scraped from the plates in cold PBS and pelleted by centrifugation at 1,000g for 2 minutes. Pellets were frozen at -80oC until needed. Cells were then lysed on ice with occasional vortexing in 1ml of lysis buffer (50mM Tris-HCl pH 7.4, 100mM NaCl, 1mM MgCl2, 0.1 mM CaCl2, 1% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS) containing 1X protease inhibitors (Roche #88665) and RNaseOUT (Invitrogen #10777019) at 4ul/ml final concentration. Next, TurboDNase (Invitrogen #AM2238, 10U), RNase A (0.13ug) and RNase T1 (0.13U) were added and samples were incubated at 37oC for 5 minutes with occasional mixing. Samples were immediately placed on ice and then centrifuged at 16,160g at 4oC for 20 minutes to clear lysate. 25ul of Protein-G Dynabeads (Invitrogen #10004D) were used per IP. Dynabeads were pre-washed with lysis buffer and pre- incubated with 3ul of Wako Anti-Mouse-Ago2 (2D4) antibody. The dynabead/antibody mixture was added to the lysate and rocked for 2 hours at 4oC. All steps after the IP were done on bead until samples were loaded into the polyacrylamide gel. -

Under Serratia Marcescens Treatment Kai Feng1,2, Xiaoyu Lu1,2, Jian Luo1,2 & Fang Tang1,2*
www.nature.com/scientificreports OPEN SMRT sequencing of the full‑length transcriptome of Odontotermes formosanus (Shiraki) under Serratia marcescens treatment Kai Feng1,2, Xiaoyu Lu1,2, Jian Luo1,2 & Fang Tang1,2* Odontotermes formosanus (Shiraki) is an important pest in the world. Serratia marcescens have a high lethal efect on O. formosanus, but the specifc insecticidal mechanisms of S. marcescens on O. formosanus are unclear, and the immune responses of O. formosanus to S. marcescens have not been clarifed. At present, genetic database resources of O. formosanus are extremely scarce. Therefore, using O. formosanus workers infected by S. marcescens and the control as experimental materials, a full-length transcriptome was sequenced using the PacBio Sequel sequencing platform. A total of 10,364 isoforms were obtained as the fnal transcriptome. The unigenes were further annotated with the Nr, Swiss-Prot, EuKaryotic Orthologous Groups (KOG), Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog public databases. In a comparison between the control group and a Serratia marcescens-infected group, a total of 259 diferentially expressed genes (DEGs) were identifed, including 132 upregulated and 127 downregulated genes. Pathway enrichment analysis indicated that the expression of the mitogen-activated protein kinase (MAPK) pathway, oxidative stress genes and the AMP-activated protein kinase (AMPK) pathway in O. formosanus may be associated with S. marcescens treatment. This research intensively studied O. formosanus at the high-throughput full-length transcriptome level, laying a foundation for further development of molecular markers and mining of target genes in this species and thereby promoting the biological control of O. -
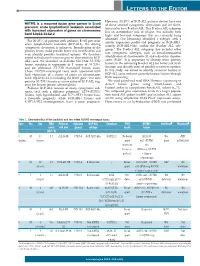
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene.