Whole Exome Sequencing in ADHD Trios from Single and Multi-Incident Families Implicates New Candidate Genes and Highlights Polygenic Transmission
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Investigation of Copy Number Variations on Chromosome 21 Detected by Comparative Genomic Hybridization
Li et al. Molecular Cytogenetics (2018) 11:42 https://doi.org/10.1186/s13039-018-0391-3 RESEARCH Open Access Investigation of copy number variations on chromosome 21 detected by comparative genomic hybridization (CGH) microarray in patients with congenital anomalies Wenfu Li, Xianfu Wang and Shibo Li* Abstract Background: The clinical features of Down syndrome vary among individuals, with those most common being congenital heart disease, intellectual disability, developmental abnormity and dysmorphic features. Complex combination of Down syndrome phenotype could be produced by partially copy number variations (CNVs) on chromosome 21 as well. By comparing individual with partial CNVs of chromosome 21 with other patients of known CNVs and clinical phenotypes, we hope to provide a better understanding of the genotype-phenotype correlation of chromosome 21. Methods: A total of 2768 pediatric patients sample collected at the Genetics Laboratory at Oklahoma University Health Science Center were screened using CGH Microarray for CNVs on chromosome 21. Results: We report comprehensive clinical and molecular descriptions of six patients with microduplication and seven patients with microdeletion on the long arm of chromosome 21. Patients with microduplication have varied clinical features including developmental delay, microcephaly, facial dysmorphic features, pulmonary stenosis, autism, preauricular skin tag, eye pterygium, speech delay and pain insensitivity. We found that patients with microdeletion presented with developmental delay, microcephaly, intrauterine fetal demise, epilepsia partialis continua, congenital coronary anomaly and seizures. Conclusion: Three patients from our study combine with four patients in public database suggests an association between 21q21.1 microduplication of CXADR gene and patients with developmental delay. One patient with 21q22.13 microdeletion of DYRK1A shows association with microcephaly and scoliosis. -

Association of the PSRC1 Rs599839 Variant with Coronary Artery Disease in a Mexican Population
medicina Communication Association of the PSRC1 rs599839 Variant with Coronary Artery Disease in a Mexican Population Martha Eunice Rodríguez-Arellano 1, Jacqueline Solares-Tlapechco 1, Paula Costa-Urrutia 1 , Helios Cárdenas-Hernández 1, Marajael Vallejo-Gómez 1, Julio Granados 2 and Sergio Salas-Padilla 3,* 1 Laboratorio de Medicina Genómica, Hospital Regional Lic. Adolfo López Mateos ISSSTE, Ciudad de Mexico 01030, Mexico; [email protected] (M.E.R.-A.); [email protected] (J.S.-T.); [email protected] (P.C.-U.); [email protected] (H.C.-H.); [email protected] (M.V.-G.) 2 División de Inmunogenética, Departamento de Trasplantes, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Ciudad de Mexico 14080, Mexico; [email protected] 3 Servicio de Cardiología, Hospital Regional Lic. Adolfo López Mateos ISSSTE, Ciudad de Mexico 01030, Mexico * Correspondence: [email protected]; Tel.: +52-555-322-2200 Received: 3 July 2020; Accepted: 12 August 2020; Published: 26 August 2020 Abstract: Background and Objectives: Coronary artery disease (CAD) is a major health problem in México. The identification of modifiable risk factors and genetic biomarkers is crucial for an integrative and personalized CAD risk evaluation. In this work, we aimed to validate in a Mexican population a set of eight selected polymorphisms previously associated with CAD, myocardial infarction (MI), or dyslipidemia. Materials and Methods: A sample of 907 subjects (394 CAD cases and 513 controls) 40–80 years old was genotyped for eight loci: PSRC1 (rs599839), MRAS (rs9818870), BTN2A1 (rs6929846), MTHFD1L (rs6922269), CDKN2B (rs1333049), KIAA1462 (rs3739998), CXCL12 (rs501120), and HNF1A (rs2259816). The association between single nucleotide polymorphisms (SNPs) and CAD was evaluated by logistic regression models. -
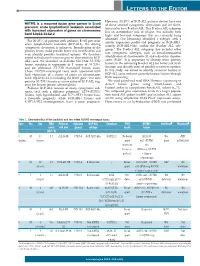
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

Viewed and Published Immediately Upon Acceptance Cited in Pubmed and Archived on Pubmed Central Yours — You Keep the Copyright
BMC Genomics BioMed Central Research article Open Access Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice Claudia Prinzen1, Dietrich Trümbach2, Wolfgang Wurst2, Kristina Endres1, Rolf Postina1 and Falk Fahrenholz*1 Address: 1Johannes Gutenberg-University, Institute of Biochemistry, Mainz, Johann-Joachim-Becherweg 30, 55128 Mainz, Germany and 2Helmholtz Zentrum München – German Research Center for Environmental Health, Institute for Developmental Genetics, Ingolstädter Landstraße 1, 85764 Neuherberg, Germany Email: Claudia Prinzen - [email protected]; Dietrich Trümbach - [email protected]; Wolfgang Wurst - [email protected]; Kristina Endres - [email protected]; Rolf Postina - [email protected]; Falk Fahrenholz* - [email protected] * Corresponding author Published: 5 February 2009 Received: 19 June 2008 Accepted: 5 February 2009 BMC Genomics 2009, 10:66 doi:10.1186/1471-2164-10-66 This article is available from: http://www.biomedcentral.com/1471-2164/10/66 © 2009 Prinzen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In a transgenic mouse model of Alzheimer disease (AD), cleavage of the amyloid precursor protein (APP) by the α-secretase ADAM10 prevented amyloid plaque formation, and alleviated cognitive deficits. Furthermore, ADAM10 overexpression increased the cortical synaptogenesis. These results suggest that upregulation of ADAM10 in the brain has beneficial effects on AD pathology. Results: To assess the influence of ADAM10 on the gene expression profile in the brain, we performed a microarray analysis using RNA isolated from brains of five months old mice overexpressing either the α-secretase ADAM10, or a dominant-negative mutant (dn) of this enzyme. -

Title CNV Analysis in Tourette Syndrome Implicates Large Genomic Rearrangements in COL8A1 and NRXN1 Author(S)
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by HKU Scholars Hub CNV Analysis in Tourette Syndrome Implicates Large Genomic Title Rearrangements in COL8A1 and NRXN1 Nag, A; Bochukova, EG; Kremeyer, B; Campbell, DD; et al.,; Author(s) Ruiz-Linares, A Citation PLoS ONE, 2013, v. 8 n. 3 Issued Date 2013 URL http://hdl.handle.net/10722/189374 Rights Creative Commons: Attribution 3.0 Hong Kong License CNV Analysis in Tourette Syndrome Implicates Large Genomic Rearrangements in COL8A1 and NRXN1 Abhishek Nag1, Elena G. Bochukova2, Barbara Kremeyer1, Desmond D. Campbell1, Heike Muller1, Ana V. Valencia-Duarte3,4, Julio Cardona5, Isabel C. Rivas5, Sandra C. Mesa5, Mauricio Cuartas3, Jharley Garcia3, Gabriel Bedoya3, William Cornejo4,5, Luis D. Herrera6, Roxana Romero6, Eduardo Fournier6, Victor I. Reus7, Thomas L Lowe7, I. Sadaf Farooqi2, the Tourette Syndrome Association International Consortium for Genetics, Carol A. Mathews7, Lauren M. McGrath8,9, Dongmei Yu9, Ed Cook10, Kai Wang11, Jeremiah M. Scharf8,9,12, David L. Pauls8,9, Nelson B. Freimer13, Vincent Plagnol1, Andre´s Ruiz-Linares1* 1 UCL Genetics Institute, Department of Genetics, Evolution and Environment, University College London, London, United Kingdom, 2 University of Cambridge Metabolic Research Laboratories, Institute of Metabolic Science, Addenbrooke’s Hospital, Cambridge, United Kingdom, 3 Laboratorio de Gene´tica Molecular, SIU, Universidad de Antioquia, Medellı´n, Colombia, 4 Escuela de Ciencias de la Salud, Universidad Pontificia -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

Molecular Mechanisms in Muscular Dystrophy : a Gene Expression Profiling Study Turk, R
Molecular mechanisms in muscular dystrophy : a gene expression profiling study Turk, R. Citation Turk, R. (2006, September 27). Molecular mechanisms in muscular dystrophy : a gene expression profiling study. Retrieved from https://hdl.handle.net/1887/4577 Version: Corrected Publisher’s Version Licence agreement concerning inclusion of doctoral License: thesis in the Institutional Repository of the University of Leiden Downloaded from: https://hdl.handle.net/1887/4577 Note: To cite this publication please use the final published version (if applicable). Molecular Mechanisms In Muscular Dystrophy A Gene Expression Profiling Study Molecular Mechanisms In Muscular Dystrophy A Gene Expression Profiling Study Proefschrift ter verkrijging van de graad van Doctor aan de Universiteit Leiden, op gezag van de Rector Magnificus Dr. D.D.Breimer, hoogleraar in de faculteit der Wiskunde en Natuurwetenschappen en die der Geneeskunde, volgens besluit van het College voor Promoties te verdedigen op woensdag 27 september 2006 klokke 15.00 uur door Rolf Turk Geboren te Leiden in 1975 Promotiecommissie Promotor Prof. Dr. G.J.B. van Ommen Co-promotores Dr. J.T. den Dunnen Dr. P.A.C. ‘t Hoen Referent Prof. Dr. R.M.W. Hofstra (Rijksuniversiteit Groningen) Overige leden Prof. Dr. M. Koenig (Université Louis Pasteur de Strasbourg) An experiment is a question which science poses to Nature, and a measurement is the recording of Nature’s answer. Max Planck Aan Maaike, Gerard en Annet Printed by: Drukkerij Duineveld ISBN-10: 90-9021042-3 ISBN-13: 978-90-9021042-1 Turk, Rolf Molecular mechanisms in muscular dystrophy. A gene expression profiling study. Thesis, Leiden University Medical Center September 27, 2006 © Rolf Turk No part of this thesis may be reproduced or transmitted in any form or by any means, without the written permission of the copyright owner Molecular Mechanisms In Muscular Dystrophies Preface 9 Chapter 1 Introduction 11 1. -

UC Irvine UC Irvine Previously Published Works
UC Irvine UC Irvine Previously Published Works Title Analysis of copy number variation in Alzheimer's disease in a cohort of clinically characterized and neuropathologically verified individuals. Permalink https://escholarship.org/uc/item/0wh7t3r9 Journal PloS one, 7(12) ISSN 1932-6203 Authors Swaminathan, Shanker Huentelman, Matthew J Corneveaux, Jason J et al. Publication Date 2012 DOI 10.1371/journal.pone.0050640 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Analysis of Copy Number Variation in Alzheimer’s Disease in a Cohort of Clinically Characterized and Neuropathologically Verified Individuals Shanker Swaminathan1,2, Matthew J. Huentelman3,4, Jason J. Corneveaux3,4, Amanda J. Myers5,6, Kelley M. Faber2, Tatiana Foroud1,2,7, Richard Mayeux8, Li Shen1,7, Sungeun Kim1,7, Mari Turk3,4, John Hardy9, Eric M. Reiman3,4,10, Andrew J. Saykin1,2,7*, for the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and the NIA-LOAD/NCRAD Family Study Group 1 Center for Neuroimaging, Department of Radiology and Imaging Sciences, Indiana University School of Medicine, Indianapolis, Indiana, United States of America, 2 Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, Indiana, United States of America, 3 Neurogenomics Division, The Translational Genomics Research Institute (TGen), Phoenix, Arizona, United States of America, 4 The Arizona Alzheimer’s Consortium, Phoenix, Arizona, United States of America, 5 Departments of Psychiatry and Behavioral Sciences, and Human Genetics and Genomics, University of Miami, Miller School of Medicine, Miami, Florida, United States of America, 6 Johnnie B. Byrd Sr. -

Presentación De Powerpoint
P5-12-03 GENOME COPY NUMBER ENTROPY AS PREDICTOR OF RESPONSE FOR NEOADJUVANT THERAPY IN EARLY BREAST CANCER Emilio Alba1,2,15, Oscar M. Rueda3, Ana Lluch2,4,15, Joan Albanell2,5, Suet-Feung Chin3, Jose Ignacio Chacón López-Muñiz2,6, Lourdes Calvo2,7, Juan de la Haba-Rodriguez2,8,15, Begoña Bermejo2,4,15, Nuria Ribelles1, Pedro Sánchez Rovira2,9, Arrate Plazaola2,10, San Antonio Breast Agustí Barnadas2,11, Beatriz Cirauqui2,12, Manuel Ramos Vázquez2,13, Angels Arcusa2,14, Eva Carrasco2, Jesús Herranz2, Massimo Chiesa2, Rosalía Caballero2, Ángela Santonja1,3, Federico Rojo2,15,16, Carlos Caldas3 1Instituto de Investigación Biomédica de Málaga (IBIMA) - Hospital Clínico Universitario Virgen de la Victoria, Málaga, Spain, 2GEICAM Spanish Breast Cancer Group, San Sebastián de los Reyes, Madrid, Spain, 3Cancer Research UK Cambridge Institute, Li Ka Shing Centre, University of Cambridge, Cancer Symposium Robinson Way, Cambridge CB2 0RE, UK, 4Hospital Clínico Universitario de Valencia, Valencia, Spain, 5Hospital del Mar, Barcelona, Spain, 6Hospital Virgen de la Salud, Toledo, Spain, 7Complejo Hospitalario Universitario de A Coruña, A Coruña, Spain, 8Instituto Maimónides de Investigación Biomédica de Córdoba (IMIBIC)–H. Universitario Reina Sofía, Universidad de Córdoba, Córdoba, Spain, 9Complejo Hospitalario de Jaén, Jaén, Spain, 10Onkologikoa, San Sebastián, Spain, 11Hospital de la Santa Creu i Sant Pau, Barcelona, Spain, 12Hospital Germans Trias i Pujol, Barcelona, Spain, 13Centro December 4-8, 2018 Oncológico de Galicia, A Coruña, Spain, 14Consorci -

Strand Breaks for P53 Exon 6 and 8 Among Different Time Course of Folate Depletion Or Repletion in the Rectosigmoid Mucosa
SUPPLEMENTAL FIGURE COLON p53 EXONIC STRAND BREAKS DURING FOLATE DEPLETION-REPLETION INTERVENTION Supplemental Figure Legend Strand breaks for p53 exon 6 and 8 among different time course of folate depletion or repletion in the rectosigmoid mucosa. The input of DNA was controlled by GAPDH. The data is shown as ΔCt after normalized to GAPDH. The higher ΔCt the more strand breaks. The P value is shown in the figure. SUPPLEMENT S1 Genes that were significantly UPREGULATED after folate intervention (by unadjusted paired t-test), list is sorted by P value Gene Symbol Nucleotide P VALUE Description OLFM4 NM_006418 0.0000 Homo sapiens differentially expressed in hematopoietic lineages (GW112) mRNA. FMR1NB NM_152578 0.0000 Homo sapiens hypothetical protein FLJ25736 (FLJ25736) mRNA. IFI6 NM_002038 0.0001 Homo sapiens interferon alpha-inducible protein (clone IFI-6-16) (G1P3) transcript variant 1 mRNA. Homo sapiens UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 15 GALNTL5 NM_145292 0.0001 (GALNT15) mRNA. STIM2 NM_020860 0.0001 Homo sapiens stromal interaction molecule 2 (STIM2) mRNA. ZNF645 NM_152577 0.0002 Homo sapiens hypothetical protein FLJ25735 (FLJ25735) mRNA. ATP12A NM_001676 0.0002 Homo sapiens ATPase H+/K+ transporting nongastric alpha polypeptide (ATP12A) mRNA. U1SNRNPBP NM_007020 0.0003 Homo sapiens U1-snRNP binding protein homolog (U1SNRNPBP) transcript variant 1 mRNA. RNF125 NM_017831 0.0004 Homo sapiens ring finger protein 125 (RNF125) mRNA. FMNL1 NM_005892 0.0004 Homo sapiens formin-like (FMNL) mRNA. ISG15 NM_005101 0.0005 Homo sapiens interferon alpha-inducible protein (clone IFI-15K) (G1P2) mRNA. SLC6A14 NM_007231 0.0005 Homo sapiens solute carrier family 6 (neurotransmitter transporter) member 14 (SLC6A14) mRNA. -

Autoantibodies As Potential Biomarkers in Breast Cancer
biosensors Review Autoantibodies as Potential Biomarkers in Breast Cancer Jingyi Qiu, Bailey Keyser, Zuan-Tao Lin and Tianfu Wu * ID Department of Biomedical Engineering, University of Houston, 3517 Cullen BLVD, SERC 2008, Houston, TX 77204, USA; [email protected] (J.Q.); [email protected] (B.K.); [email protected] (Z.-T.L.) * Correspondence: [email protected]; Tel.: +1-713-743-0142 Received: 14 June 2018; Accepted: 11 July 2018; Published: 13 July 2018 Abstract: Breast cancer is a major cause of mortality in women; however, technologies for early stage screening and diagnosis (e.g., mammography and other imaging technologies) are not optimal for the accurate detection of cancer. This creates demand for a more effective diagnostic means to replace or be complementary to existing technologies for early discovery of breast cancer. Cancer neoantigens could reflect tumorigenesis, but they are hardly detectable at the early stage. Autoantibodies, however, are biologically amplified and hence may be measurable early on, making them promising biomarkers to discriminate breast cancer from healthy tissue accurately. In this review, we summarized the recent findings of breast cancer specific antigens and autoantibodies, which may be useful in early detection, disease stratification, and monitoring of treatment responses of breast cancer. Keywords: autoantibody; breast cancer; early diagnosis; immunotherapy 1. Introduction Breast cancer is the prevailing cancer among women in developing and developed countries [1]. As such, screening and early diagnosis with respect to risk stratification are critical for prevention and early intervention of the disease, leading to better therapeutic outcomes [2,3]. Breast cancer itself is genetically heterogeneous and expresses a variety of aberrant proteins that, until recently, were un-utilizable.