Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Acteoside from Ligustrum Robustum (Roxb.) Blume Ameliorates Lipid Metabolism and Synthesis in a Hepg2 Cell Model of Lipid Accumulation
ORIGINAL RESEARCH published: 24 May 2019 doi: 10.3389/fphar.2019.00602 Acteoside From Ligustrum robustum (Roxb.) Blume Ameliorates Lipid Metabolism and Synthesis in a HepG2 Cell Model of Lipid Accumulation Le Sun 1,2†, Fan Yu 1,2†, Fan Yi 3, Lijia Xu 1,2*, Baoping Jiang 1,2*, Liang Le 1,2 and Peigen Xiao 1,2 1 Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, China, 2 Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Beijing, China, 3 Key Laboratory of Cosmetics, China National Light Industry, Beijing Technology and Business Edited by: University, Beijing, China Min Ye, Peking University, China We aimed to ascertain the mechanism underlying the effects of acteoside (ACT) from Reviewer: Wei Song, Ligustrum robustum (Roxb.) Blume (Oleaceae) on lipid metabolism and synthesis. ACT, Peking Union Medical College a water-soluble phenylpropanoid glycoside, is the most abundant and major active Hospital (CAMS), component of L. robustum; the leaves of L. robustum, known as kudingcha (bitter tea), China Shuai Ji, have long been used in China as an herbal tea for weight loss. Recently, based on previous Xuzhou Medical University, studies, our team reached a preliminary conclusion that phenylpropanoid glycosides from China L. robustum most likely contribute substantially to reducing lipid levels, but the mechanism *Correspondence: Lijia Xu remains unclear. Here, we conducted an in silico screen of currently known phenylethanoid [email protected] glycosides from L. robustum and attempted to explore the hypolipidemic mechanism of Baoping Jiang ACT, the representative component of phenylethanoid glycosides in L. -

Evolution, Dynamic Expression Changes and Regulatory Characteristics of Gene Families Involved in the Glycerophosphate Pathway O
www.nature.com/scientificreports OPEN Evolution, dynamic expression changes and regulatory characteristics of gene families Received: 2 October 2018 Accepted: 14 August 2019 involved in the glycerophosphate Published: xx xx xxxx pathway of triglyceride synthesis in chicken (Gallus gallus) Liyu Yang1, Ziming Liu1, Kepeng Ou2, Taian Wang1, Zhuanjian Li1,3,4, Yadong Tian1,3,4, Yanbin Wang1,3,4, Xiangtao Kang1,3,4, Hong Li1,3,4 & Xiaojun Liu1,3,4 It is well documented that four gene families, including the glycerophosphate acyltransferases (GPATs), acylglycerophosphate acyltransferases (AGPATs), lipid phosphate phosphohydrolases (LPINs) and diacylglycerol acyltransferases (DGATs), are involved in the glycerophosphate pathway of de novo triglyceride (TG) biosynthesis in mammals. However, no systematic analysis has been conducted to characterize the gene families in poultry. In this study, the sequences of gene family members in the glycerophosphate pathway were obtained by screening the public databases. The phylogenetic tree, gene structures and conserved motifs of the corresponding proteins were evaluated. Dynamic expression changes of the genes at diferent developmental stages were analyzed by qRT-PCR. The regulatory characteristics of the genes were analyzed by in vivo experiments. The results showed that the GPAT, AGPAT and LPIN gene families have 2, 7 and 2 members, respectively, and they were classifed into 2, 4 and 2 cluster respectively based on phylogenetic analysis. All of the genes except AGPAT1 were extensively expressed in various tissues. Estrogen induction upregulated the expression of GPAM and AGPAT2, downregulated the expression of AGPAT3, AGPAT9, LPIN1 and LPIN2, and had no efect on the expression of the other genes. These fndings provide a valuable resource for further investigation of lipid metabolism in liver of chicken. -

ACAT) in Cholesterol Metabolism: from Its Discovery to Clinical Trials and the Genomics Era
H OH metabolites OH Review Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era Qimin Hai and Jonathan D. Smith * Department of Cardiovascular & Metabolic Sciences, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-444-2248 Abstract: The purification and cloning of the acyl-coenzyme A: cholesterol acyltransferase (ACAT) enzymes and the sterol O-acyltransferase (SOAT) genes has opened new areas of interest in cholesterol metabolism given their profound effects on foam cell biology and intestinal lipid absorption. The generation of mouse models deficient in Soat1 or Soat2 confirmed the importance of their gene products on cholesterol esterification and lipoprotein physiology. Although these studies supported clinical trials which used non-selective ACAT inhibitors, these trials did not report benefits, and one showed an increased risk. Early genetic studies have implicated common variants in both genes with human traits, including lipoprotein levels, coronary artery disease, and Alzheimer’s disease; however, modern genome-wide association studies have not replicated these associations. In contrast, the common SOAT1 variants are most reproducibly associated with testosterone levels. Keywords: cholesterol esterification; atherosclerosis; ACAT; SOAT; inhibitors; clinical trial Citation: Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its 1. Introduction Discovery to Clinical Trials and the The acyl-coenzyme A:cholesterol acyltransferase (ACAT; EC 2.3.1.26) enzyme family Genomics Era. Metabolites 2021, 11, consists of membrane-spanning proteins, which are primarily located in the endoplasmic 543. https://doi.org/10.3390/ reticulum [1]. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Whole Exome Sequencing in ADHD Trios from Single and Multi-Incident Families Implicates New Candidate Genes and Highlights Polygenic Transmission
European Journal of Human Genetics (2020) 28:1098–1110 https://doi.org/10.1038/s41431-020-0619-7 ARTICLE Whole exome sequencing in ADHD trios from single and multi-incident families implicates new candidate genes and highlights polygenic transmission 1,2 1 1,2 1,3 1 Bashayer R. Al-Mubarak ● Aisha Omar ● Batoul Baz ● Basma Al-Abdulaziz ● Amna I. Magrashi ● 1,2 2 2,4 2,4,5 6 Eman Al-Yemni ● Amjad Jabaan ● Dorota Monies ● Mohamed Abouelhoda ● Dejene Abebe ● 7 1,2 Mohammad Ghaziuddin ● Nada A. Al-Tassan Received: 26 September 2019 / Revised: 26 February 2020 / Accepted: 10 March 2020 / Published online: 1 April 2020 © The Author(s) 2020. This article is published with open access Abstract Several types of genetic alterations occurring at numerous loci have been described in attention deficit hyperactivity disorder (ADHD). However, the role of rare single nucleotide variants (SNVs) remains under investigated. Here, we sought to identify rare SNVs with predicted deleterious effect that may contribute to ADHD risk. We chose to study ADHD families (including multi-incident) from a population with a high rate of consanguinity in which genetic risk factors tend to 1234567890();,: 1234567890();,: accumulate and therefore increasing the chance of detecting risk alleles. We employed whole exome sequencing (WES) to interrogate the entire coding region of 16 trios with ADHD. We also performed enrichment analysis on our final list of genes to identify the overrepresented biological processes. A total of 32 rare variants with predicted damaging effect were identified in 31 genes. At least two variants were detected per proband, most of which were not exclusive to the affected individuals. -

Actin Binding LIM 1 (Ablim1) Negatively Controls Osteoclastogenesis by Regulating Cell Migration and Fusion
Received: 14 September 2017 | Accepted: 16 March 2018 DOI: 10.1002/jcp.26605 ORIGINAL RESEARCH ARTICLE Actin binding LIM 1 (abLIM1) negatively controls osteoclastogenesis by regulating cell migration and fusion Haruna Narahara1,2 | Eiko Sakai1 | Yu Yamaguchi1 | Shun Narahara1 | Mayumi Iwatake1,* | Kuniaki Okamoto1,† | Noriaki Yoshida2 | Takayuki Tsukuba1 1 Department of Dental Pharmacology, Graduate School of Biomedical Sciences, Actin binding LIM 1 (abLIM1) is a cytoskeletal actin-binding protein that has been Nagasaki University, Nagasaki, Japan implicated in interactions between actin filaments and cytoplasmic targets. Previous 2 Department of Orthodontics and Dentofacial Orthopedics, Graduate School of Biomedical biochemical and cytochemical studies have shown that abLIM1 interacts and co- Sciences, Nagasaki University, Nagasaki, Japan localizes with F-actin in the retina and muscle. However, whether abLIM1 regulates Correspondence osteoclast differentiation has not yet been elucidated. In this study, we examined the Takayuki Tsukuba, Department of Dental role of abLIM1 in osteoclast differentiation and function. We found that abLIM1 Pharmacology, Graduate School of Biomedical Sciences, Nagasaki University, Nagasaki 852- expression was upregulated during receptor activator of nuclear factor kappa-B ligand 8588, Japan. (RANKL)-induced osteoclast differentiation, and that a novel transcript of abLIM1 was Email: [email protected] exclusively expressed in osteoclasts. Overexpression of abLIM1 in the murine Funding information monocytic cell line, RAW-D suppressed osteoclast differentiation and decreased JSPS KAKENHI, Grant numbers: 15H05298, 16K15790, 17H04379 expression of several osteoclast-marker genes. By contrast, small interfering RNA-induced knockdown of abLIM1 enhanced the formation of multinucleated osteoclasts and markedly increased the expression of the osteoclast-marker genes. Mechanistically, abLIM1 regulated the localization of tubulin, migration, and fusion in osteoclasts. -

Functional Significance of the Two ACOX1 Isoforms and Their
Laboratory Investigation (2010) 90, 696–708 & 2010 USCAP, Inc All rights reserved 0023-6837/10 $32.00 Functional significance of the two ACOX1 isoforms and their crosstalks with PPARa and RXRa Aurore Vluggens1,2,3, Pierre Andreoletti1,2, Navin Viswakarma3, Yuzhi Jia3, Kojiro Matsumoto3, Wim Kulik4, Mushfiquddin Khan5, Jiansheng Huang3, Dongsheng Guo3, Sangtao Yu3, Joy Sarkar3, Inderjit Singh5, M Sambasiva Rao3, Ronald J Wanders4, Janardan K Reddy3 and Mustapha Cherkaoui-Malki1,2 Disruption of the peroxisomal acyl-CoA oxidase 1 (Acox1) gene in the mouse results in the development of severe microvesicular hepatic steatosis and sustained activation of peroxisome proliferator-activated receptor-a (PPARa). These mice manifest spontaneous massive peroxisome proliferation in regenerating hepatocytes and eventually develop hepatocellular carcinomas. Human ACOX1, the first and rate-limiting enzyme of the peroxisomal b-oxidation pathway, has two isoforms including ACOX1a and ACOX1b, transcribed from a single gene. As ACOX1a shows reduced activity toward palmitoyl-CoA as compared with ACOX1b, we used adenovirally driven ACOX1a and ACOX1b to investigate their efficacy in the reversal of hepatic phenotype in Acox1(À/À) mice. In this study, we show that human ACOX1b is markedly effective in reversing the ACOX1 null phenotype in the mouse. In addition, expression of human ACOX1b was found to restore the production of nervonic (24:1) acid and had a negative impact on the recruitment of coactivators to the PPARa-response unit, which suggests that nervonic acid might well be an endogenous PPARa antagonist, with nervonoyl-CoA probably being the active form of nervonic acid. In contrast, restoration of docosahexaenoic (22:6) acid level, a retinoid-X-receptor (RXRa) agonist, was dependent on the concomitant hepatic expression of both ACOX1a and ACOX1b isoforms. -
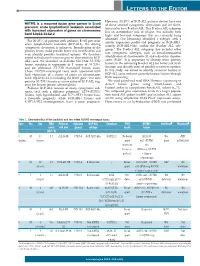
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Original Article Interactions of ABLIMI and CXCL5 with Mirnas As a Prognostic Indicator for Clinical Outcome of Osteosarcoma
Int J Clin Exp Med 2016;9(8):15345-15353 www.ijcem.com /ISSN:1940-5901/IJCEM0029430 Original Article Interactions of ABLIMI and CXCL5 with miRNAs as a prognostic indicator for clinical outcome of osteosarcoma Pengfei Gao1, Zhaowei Teng2, lixin Ji1, Wengui Xie1 1Department of Spinal Surgery, North Medical District of Linyi People’s Hospital Group, Linyi, China; 2Department of Orthopedic Surgery, People’s Hospital of Yuxi City, The 6th Affiliated Hospital of Kunming Medical College, Yuxi, China Received March 29, 2016; Accepted July 10, 2016; Epub August 15, 2016; Published August 30, 2016 Abstract: Osteosarcoma (OS) is a kind of high-grade bone-forming malignancy with poor prognosis, causing huge economic losses to the patients and society. Identification of novel biomarkers could be beneficial for the diagnosis and prognosis of OS patients. We aimed to explore the molecular mechanism of osteosarcoma by the differentially expressed miRNAs and mRNAs screened out by microarray data together with the function and metabolic pathway analysis of target genes. Expression data of miRNA and mRNA were downloaded from The Genome Expression Omnibus (GEO) dataset. Differentially expressed miRNAs of osteosarcoma were screened out by two-fold principle. Target genes of differentially expressed miRNAs were extracted out by miRTarBase software. Besides, DAVID data- set was used for the functional annotation. Differentially expressed genes of mRNA microarray were screened out for the study of relation between miRNA and mRNA. A total of 33 differentially expressed miRNAs were screened out, including 27 up-regulated miRNAs and 6 down-regulated miRNAs. Three miRNAs, hsa-miR-182, hsa-miR-486- 5p and hsa-miR760, were found to be the most differentially expressed ones, which were all up-regulated and could be used to differentiate osteosarcoma tissues from the normal ones.