ACAT) in Cholesterol Metabolism: from Its Discovery to Clinical Trials and the Genomics Era
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acteoside from Ligustrum Robustum (Roxb.) Blume Ameliorates Lipid Metabolism and Synthesis in a Hepg2 Cell Model of Lipid Accumulation
ORIGINAL RESEARCH published: 24 May 2019 doi: 10.3389/fphar.2019.00602 Acteoside From Ligustrum robustum (Roxb.) Blume Ameliorates Lipid Metabolism and Synthesis in a HepG2 Cell Model of Lipid Accumulation Le Sun 1,2†, Fan Yu 1,2†, Fan Yi 3, Lijia Xu 1,2*, Baoping Jiang 1,2*, Liang Le 1,2 and Peigen Xiao 1,2 1 Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, China, 2 Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Beijing, China, 3 Key Laboratory of Cosmetics, China National Light Industry, Beijing Technology and Business Edited by: University, Beijing, China Min Ye, Peking University, China We aimed to ascertain the mechanism underlying the effects of acteoside (ACT) from Reviewer: Wei Song, Ligustrum robustum (Roxb.) Blume (Oleaceae) on lipid metabolism and synthesis. ACT, Peking Union Medical College a water-soluble phenylpropanoid glycoside, is the most abundant and major active Hospital (CAMS), component of L. robustum; the leaves of L. robustum, known as kudingcha (bitter tea), China Shuai Ji, have long been used in China as an herbal tea for weight loss. Recently, based on previous Xuzhou Medical University, studies, our team reached a preliminary conclusion that phenylpropanoid glycosides from China L. robustum most likely contribute substantially to reducing lipid levels, but the mechanism *Correspondence: Lijia Xu remains unclear. Here, we conducted an in silico screen of currently known phenylethanoid [email protected] glycosides from L. robustum and attempted to explore the hypolipidemic mechanism of Baoping Jiang ACT, the representative component of phenylethanoid glycosides in L. -

SOAT1 Polyclonal Antibody
PRODUCT DATA SHEET Bioworld Technology,Inc. SOAT1 polyclonal antibody Catalog: BS8121 Host: Rabbit Reactivity: Human,Mouse,Rat BackGround: Purification&Purity: SOAT1 (sterol O-acyltransferase-1), also designated The antibody was affinity-purified from rabbit antiserum ACAT1, is a homotetra-meric enzyme that catalyzes the by affinity-chromatography using epitope-specific im- formation of cholesterol esters from cholesterol and long munogen and the purity is > 95% (by SDS-PAGE). chain fatty acyl-coenzyme A (acyl-CoA). The gene en- Applications: coding human SOAT1 maps to chromosome 1 and is ex- WB 1:500 - 1:2000 pressed as a protein that localizes to the endoplasmic re- Storage&Stability: ticulum (ER) in several tissues, including liver, kidney, Store at 4°C short term. Aliquot and store at -20°C long adrenal glands and macrophages. SOAT1 is involved in term. Avoid freeze-thaw cycles. cellular cholesterol homeostasis as well as in foam cell Specificity: formation and the subsequent progression of atheroscle- SOAT1 polyclonal antibody detects endogenous levels of rosis. Several SOAT inhibitors have been developed for SOAT1 protein. the treatment of atherosclerosis. SOAT2 (sterol DATA: O-acyltransferase-2), also known as ACAT2 (acyl-CoA:cholesterol acyltransferase-2), participates in lipo-protein assembly, catalyzing cholesterol esterifica- tion in mammalian cells. SOAT2 is an integral membrane protein that localizes to the endoplasmic reticulum of human intestinal cells. SOAT2 deficiency contributes to severe mental retardation and hypotonus. Product: WesternBlot (WB) analysis of SOAT1 polyclonal antibody Rabbit IgG, 1mg/ml in PBS with 0.02% sodium azide, Note: 50% glycerol, pH7.2 For research use only, not for use in diagnostic procedure. -

Role of De Novo Cholesterol Synthesis Enzymes in Cancer Jie Yang1,2, Lihua Wang1,2, Renbing Jia1,2
Journal of Cancer 2020, Vol. 11 1761 Ivyspring International Publisher Journal of Cancer 2020; 11(7): 1761-1767. doi: 10.7150/jca.38598 Review Role of de novo cholesterol synthesis enzymes in cancer Jie Yang1,2, Lihua Wang1,2, Renbing Jia1,2 1. Department of Ophthalmology, Ninth People’s Hospital of Shanghai, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 2. Shanghai Key Laboratory of Orbital Diseases and Ocular Oncology, Shanghai, China. Corresponding authors: Renbing Jia, [email protected] and Lihua Wang, [email protected]. Department of Ophthalmology, Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, No. 639 Zhi Zao Ju Road, Shanghai 200011, China © The author(s). This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See http://ivyspring.com/terms for full terms and conditions. Received: 2019.07.21; Accepted: 2019.11.30; Published: 2020.01.17 Abstract Despite extensive research in the cancer field, cancer remains one of the most prevalent diseases. There is an urgent need to identify specific targets that are safe and effective for the treatment of cancer. In recent years, cancer metabolism has come into the spotlight in cancer research. Lipid metabolism, especially cholesterol metabolism, plays a critical role in membrane synthesis as well as lipid signaling in cancer. This review focuses on the contribution of the de novo cholesterol synthesis pathway to tumorigenesis, cancer progression and metastasis. In conclusion, cholesterol metabolism could be an effective target for novel anticancer treatment. Key words: metabolic reprogramming, de novo cholesterol synthesis, cancer progress Introduction Over the past few decades, numerous published that cholesterol plays a critical role in cancer studies have focused on cancer cell metabolism and progression15-19. -
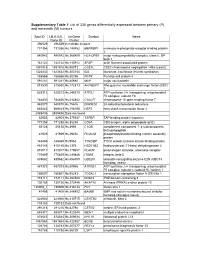
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

High Throughput Computational Mouse Genetic Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.09.01.278465; this version posted January 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. High Throughput Computational Mouse Genetic Analysis Ahmed Arslan1+, Yuan Guan1+, Zhuoqing Fang1, Xinyu Chen1, Robin Donaldson2&, Wan Zhu, Madeline Ford1, Manhong Wu, Ming Zheng1, David L. Dill2* and Gary Peltz1* 1Department of Anesthesia, Stanford University School of Medicine, Stanford, CA; and 2Department of Computer Science, Stanford University, Stanford, CA +These authors contributed equally to this paper & Current Address: Ecree Durham, NC 27701 *Address correspondence to: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.09.01.278465; this version posted January 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Background: Genetic factors affecting multiple biomedical traits in mice have been identified when GWAS data that measured responses in panels of inbred mouse strains was analyzed using haplotype-based computational genetic mapping (HBCGM). Although this method was previously used to analyze one dataset at a time; but now, a vast amount of mouse phenotypic data is now publicly available, which could lead to many more genetic discoveries. Results: HBCGM and a whole genome SNP map covering 53 inbred strains was used to analyze 8462 publicly available datasets of biomedical responses (1.52M individual datapoints) measured in panels of inbred mouse strains. As proof of concept, causative genetic factors affecting susceptibility for eye, metabolic and infectious diseases were identified when structured automated methods were used to analyze the output. -

Human 3B-Hydroxysteroid Dehydrogenase Deficiency Seems
M-A Burckhardt and others Histology of HSD3B2 deficiency 173:5 K1–K12 Case Report Human 3b-hydroxysteroid dehydrogenase deficiency seems to affect fertility but may not harbor a tumor risk: lesson from an experiment of nature Marie-Anne Burckhardt1, Sameer S Udhane1,†, Nesa Marti1,†, Isabelle Schnyder2, Coya Tapia3, John E Nielsen4, Primus E Mullis1, Ewa Rajpert-De Meyts4 and Christa E Flu¨ ck1 1Pediatric Endocrinology and Diabetology, Departments of Pediatrics and Clinical Research, 2Pediatric Surgery and Correspondence 3Institute of Pathology, University of Bern, CH-3010 Bern, Switzerland and 4Department of Growth and should be addressed Reproduction, Rigshospitalet, Copenhagen University Hospital, Copenhagen, Denmark to C E Flu¨ ck †S S Udhane and N Marti are now at Graduate School Bern, University of Bern, Bern Switzerland Email christa.fl[email protected] Abstract Context:3b-hydroxysteroid dehydrogenase deficiency (3bHSD) is a rare disorder of sexual development and steroidogenesis. There are two isozymes of 3bHSD, HSD3B1 and HSD3B2. Human mutations are known for the HSD3B2 gene which is expressed in the gonads and the adrenals. Little is known about testis histology, fertility and malignancy risk. Objective: To describe the molecular genetics, the steroid biochemistry, the (immuno-)histochemistry and the clinical implications of a loss-of-function HSD3B2 mutation. Methods: Biochemical, genetic and immunohistochemical investigations on human biomaterials. Results: A 46,XY boy presented at birth with severe undervirilization of the external genitalia. Steroid profiling showed low steroid production for mineralocorticoids, glucocorticoids and sex steroids with typical precursor metabolites for HSD3B2 European Journal of Endocrinology deficiency. The genetic analysis of the HSD3B2 gene revealed a homozygous c.687del27 deletion. -

Genotyping S328F SNP of Human ACAT2 Among CVD Patients and Normal Population from North Western India and Its Associated Lipid Profile P Balgir, G Kaur, Divya
The Internet Journal of Genomics and Proteomics ISPUB.COM Volume 4 Number 2 Genotyping S328F SNP of human ACAT2 among CVD patients and normal population from North Western India and its associated lipid profile P Balgir, G Kaur, Divya Citation P Balgir, G Kaur, Divya. Genotyping S328F SNP of human ACAT2 among CVD patients and normal population from North Western India and its associated lipid profile. The Internet Journal of Genomics and Proteomics. 2008 Volume 4 Number 2. Abstract Acyl-coenzyme A: cholesterol acyltransferase 2 (ACAT2, EC2.3.1.26) is an integral membrane protein located in the rough endoplasmic reticulum (ER). It maintains a balance between the availability of free and esterified cholesterol and is critically important for cell function. Purpose: The present study discusses the standardization of protocol to delineate the role of SNP S328F in ACAT2 gene. Methods: SNP S328F stability prediction was done using CUPSAT and various parameters like Primer concentration, amount of taq and glycerol etc were studied to set a protocol using Polymerase Chain reaction to achieve a product of 127 base pair. Restriction digestion was done overnight using BsmA1 enzyme to study the genotypes. The present study was used to study the SNP in 200 healthy subjects and 166 CAD patient group.Results: Higher frequency of C allele was observed in CAD patients and higher levels of TC were observed to be related to the presence of allele T which is associated with the presence of hydrophobic amino acid F at position 328. Discussion and Conclusion: Presence of T allele leads to more accumulation of cholesterol esters as it increases the protein stability in the cytoplasmic region and consequently making the individual more susceptible to atherosclerosis. -

Nuclear Receptors Control Pro-Viral and Antiviral Metabolic Responses to Hepatitis C Virus Infection
ARTICLE PUBLISHED ONLINE: 10 OCTOBER 2016 | DOI: 10.1038/NCHEMBIO.2193 Nuclear receptors control pro-viral and antiviral metabolic responses to hepatitis C virus infection Gahl Levy1,2,11, Naomi Habib1–3,11, Maria Angela Guzzardi1,4,11, Daniel Kitsberg1,2, David Bomze1,2, Elishai Ezra1,5, Basak E Uygun6, Korkut Uygun6, Martin Trippler7, Joerg F Schlaak7, Oren Shibolet8, Ella H Sklan9, Merav Cohen1,2, Joerg Timm10, Nir Friedman1,2 & Yaakov Nahmias1,2* Viruses lack the basic machinery needed to replicate and therefore must hijack the host’s metabolism to propagate. Virus- induced metabolic changes have yet to be systematically studied in the context of host transcriptional regulation, and such studies shoul offer insight into host–pathogen metabolic interplay. In this work we identified hepatitis C virus (HCV)- responsive regulators by coupling system-wide metabolic-flux analysis with targeted perturbation of nuclear receptors in primary human hepatocytes. We found HCV-induced upregulation of glycolysis, ketogenesis and drug metabolism, with glycolysis controlled by activation of HNF4a, ketogenesis by PPARa and FXR, and drug metabolism by PXR. Pharmaceutical inhibition of HNF4a reversed HCV-induced glycolysis, blocking viral replication while increasing apoptosis in infected cells showing virus-induced dependence on glycolysis. In contrast, pharmaceutical inhibition of PPARa or FXR reversed HCV-induced ketogenesis but increased viral replication, demonstrating a novel host antiviral response. Our results show that virus-induced changes to a host’s metabolism can be detrimental to its life cycle, thus revealing a biologically complex relationship between virus and host. iral infection is one of the leading medical challenges of enzymes of oxidative phosphorylation as well6. -

Cmah Deficiency May Lead to Age-Related Hearing Loss by Influencing Mirna-PPAR Mediated Signaling Pathway
Cmah deficiency may lead to age-related hearing loss by influencing miRNA-PPAR mediated signaling pathway Juhong Zhang1, Na Wang2 and Anting Xu2,3 1 Department of Otolaryngology, Shanghai Jiao Tong University Affiliated Sixth People's Hospital South Campus, Southern Medical University Affiliated Fengxian Hospital, Shanghai, China 2 Department of Otolaryngology/Head and Neck Surgery, the Second Hospital of Shandong University, Jinan, China 3 NHC. Key Laboratory of Otorhinolaryngology, Shandong University, Jinan, China ABSTRACT Background. Previous evidence has indicated CMP-Neu5Ac hydroxylase (Cmah) disruption inducesaging-related hearing loss (AHL). However, its function mechanisms remain unclear. This study was to explore the mechanisms of AHL by using microarray analysis in the Cmah deficiency animal model. Methods. Microarray dataset GSE70659 was available from the Gene Expression Omnibus database, including cochlear tissues from wild-type and Cmah-null C57BL/6J mice with old age (12 months, n D 3). Differentially expressed genes (DEGs) were identified using the Linear Models for Microarray data method and a protein– protein interaction (PPI) network was constructed using data from the Search Tool for the Retrieval of Interacting Genes database followed by module analysis. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis was performed using the Database for Annotation, Visualization and Integrated Discovery. The upstream miRNAs and potential small-molecule drugs were predicted by miRwalk2.0 and Connectivity Map, respectively. Results. A total of 799 DEGs (449 upregulated and 350 downregulated) were identified. Upregulated DEGs were involved in Cell adhesion molecules (ICAM1, intercellular adhesion molecule 1) and tumor necrosis factor (TNF) signaling pathway (FOS, FBJ Submitted 22 January 2019 osteosarcoma oncogene; ICAM1), while downregulated DEGs participated in PPAR Accepted 26 March 2019 signaling pathway (PPARG, peroxisome proliferator-activated receptor gamma). -

Anti-SOAT2 / ACAT2 Antibody (ARG57814)
Product datasheet [email protected] ARG57814 Package: 250 μl anti-SOAT2 / ACAT2 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes SOAT2 / ACAT2 Tested Reactivity Hu, Ms, Rat, Pig, Sheep Tested Application ICC/IF, IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name SOAT2 / ACAT2 Antigen Species Human Immunogen Synthetic peptide around the N-terminus of Human SOAT2 / ACAT2. Conjugation Un-conjugated Alternate Names Cholesterol acyltransferase 2; ACACT2; ACAT-2; Sterol O-acyltransferase 2; ACAT2; ARGP2; Acyl- coenzyme A:cholesterol acyltransferase 2; EC 2.3.1.26 Application Instructions Application table Application Dilution ICC/IF 1:200 - 1:500 IHC-P 1:200 - 1:500 WB 1:200 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 60 kDa Properties Form Liquid Purification Affinity purification with immunogen. Buffer TBS (pH 7.4), 0.02% Sodium azide, 50% Glycerol and 0.1% BSA. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol and 0.1% BSA Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol SOAT2 Gene Full Name sterol O-acyltransferase 2 Background Summary:This gene is a member of a small family of acyl coenzyme A:cholesterol acyltransferases. -

Two Siblings with Beta-Ketothiolase Deficiency: One Genetic Defect Two
J Pediatr Res 2016;3(2):113-6 DO I: 10.4274/jpr.25338 Case Report / Olgu Sunumu Two Siblings with Beta-Ketothiolase Deficiency: One Genetic Defect Two Different Pictures Beta-Ketotiolaz Eksikliği Tanısı Alan İki Kardeş: Tek Genetik Defekt İki Farklı Tablo Melis Demir Köse1, Ebru Canda1, Mehtap Kağnıcı1, Rana İşgüder2, Aycan Ünalp3, Sema Kalkan Uçar1, Luzy Bahr4, Corinne Britschgi4, Jörn Oliver Sass4, Mahmut Çoker4 1Ege University Faculty of Medicine, Department of Pediatric Metabolism, İzmir, Turkey 2Behçet Uz Children Training and Research Hospital, Clinic of Pediatric Intensive Care, İzmir, Turkey 3Behçet Uz Children Training and Research Hospital, Clinic of Neurology, İzmir, Turkey 4Zürich University Children’s Hospital, Department of Clinical Chemistry and Biochemistry, Zürich, Switzerland ABS TRACT ÖZ Deficiency of mitochondrial acetoacetyl-coenzyme A thiolase T2 Mitokondriyal asetoasetil-koenzim A tiolaz T2 eksikliği (MAT- β-ketotiolaz) (methylacetoacetyl-coenzyme A thiolase, MAT) or β-ketothiolase is a rare ketoasidotik ataklarla karakterize nadir bir otozomal resesif hastalıktır. Klinik autosomal recessive disorder that is characterized by ketoacidosis episodes. gidişat tamamen normal gelişimsel süreçten ağır mental retardasyona hatta Outcomes vary from normal development to severe cognitive impairment or atak sonrası ölüme kadar değişebilir. Klasik T2 eksikliğinin idrar organik asit even death after an acute episode of ketoacidosis. The classical biochemical ve karnitin profiline yansıyan biyokimyasal özellikleri ACAT1 geninde her iki profile of T2 deficiency is a result of mutations in both alleles of the ACAT1 allelde de oluşacak mutasyonlar sonucu görülmektedir. Bu yazımızda oldukça gene and comprises characteristic abnormalities in urinary organic acids and farklı fenotipik özellikler gösteren ancak aynı mutasyonu paylaşan iki kardeşin blood or plasma acylcarnitine profiles. -

Genome-Wide Expression Profiling Establishes Novel Modulatory Roles
Batra et al. BMC Genomics (2017) 18:252 DOI 10.1186/s12864-017-3635-4 RESEARCHARTICLE Open Access Genome-wide expression profiling establishes novel modulatory roles of vitamin C in THP-1 human monocytic cell line Sakshi Dhingra Batra, Malobi Nandi, Kriti Sikri and Jaya Sivaswami Tyagi* Abstract Background: Vitamin C (vit C) is an essential dietary nutrient, which is a potent antioxidant, a free radical scavenger and functions as a cofactor in many enzymatic reactions. Vit C is also considered to enhance the immune effector function of macrophages, which are regarded to be the first line of defence in response to any pathogen. The THP- 1 cell line is widely used for studying macrophage functions and for analyzing host cell-pathogen interactions. Results: We performed a genome-wide temporal gene expression and functional enrichment analysis of THP-1 cells treated with 100 μM of vit C, a physiologically relevant concentration of the vitamin. Modulatory effects of vitamin C on THP-1 cells were revealed by differential expression of genes starting from 8 h onwards. The number of differentially expressed genes peaked at the earliest time-point i.e. 8 h followed by temporal decline till 96 h. Further, functional enrichment analysis based on statistically stringent criteria revealed a gamut of functional responses, namely, ‘Regulation of gene expression’, ‘Signal transduction’, ‘Cell cycle’, ‘Immune system process’, ‘cAMP metabolic process’, ‘Cholesterol transport’ and ‘Ion homeostasis’. A comparative analysis of vit C-mediated modulation of gene expression data in THP-1cells and human skin fibroblasts disclosed an overlap in certain functional processes such as ‘Regulation of transcription’, ‘Cell cycle’ and ‘Extracellular matrix organization’, and THP-1 specific responses, namely, ‘Regulation of gene expression’ and ‘Ion homeostasis’.