Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intratumoral Estrogen Disposition in Breast Cancer
Published OnlineFirst March 9, 2010; DOI: 10.1158/1078-0432.CCR-09-2481 Published Online First on March 9, 2010 as 10.1158/1078-0432.CCR-09-2481 Clinical Human Cancer Biology Cancer Research Intratumoral Estrogen Disposition in Breast Cancer Ben P. Haynes1, Anne Hege Straume3,4, Jürgen Geisler6, Roger A'Hern7, Hildegunn Helle5, Ian E. Smith2, Per E. Lønning3,5, and Mitch Dowsett1 Abstract Purpose: The concentration of estradiol (E2) in breast tumors is significantly higher than that in plas- ma, particularly in postmenopausal women. The contribution of local E2 synthesis versus uptake of E2 from the circulation is controversial. Our aim was to identify possible determinants of intratumoral E2 levels in breast cancer patients. Experimental Design: The expression of genes involved in estrogen synthesis, metabolism, and sig- naling was measured in 34 matched samples of breast tumor and normal breast tissue, and their corre- lation with estrogen concentrations assessed. Results: ESR1 (9.1-fold; P < 0.001) and HSD17B7 (3.5-fold; P < 0.001) were upregulated in ER+ tumors compared with normal tissues, whereas STS (0.34-fold; P < 0.001) and HSD17B5 (0.23-fold; P < 0.001) were downregulated. Intratumoral E2 levels showed a strong positive correlation with ESR1 expression in all patients (Spearman r = 0.55, P < 0.001) and among the subgroups of postmenopausal (r = 0.76, P < 0.001; n = 23) and postmenopausal ER+ patients (r = 0.59, P = 0.013; n = 17). HSD17B7 expression showed a significant positive correlation (r =0.59,P < 0.001) whereas HSD17B2 (r = −0.46, P = 0.0057) and HSD17B12 (r = −0.45, P = 0.0076) showed significant negative correlations with intratumoral E2 in all patients. -

ACAT) in Cholesterol Metabolism: from Its Discovery to Clinical Trials and the Genomics Era
H OH metabolites OH Review Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era Qimin Hai and Jonathan D. Smith * Department of Cardiovascular & Metabolic Sciences, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-444-2248 Abstract: The purification and cloning of the acyl-coenzyme A: cholesterol acyltransferase (ACAT) enzymes and the sterol O-acyltransferase (SOAT) genes has opened new areas of interest in cholesterol metabolism given their profound effects on foam cell biology and intestinal lipid absorption. The generation of mouse models deficient in Soat1 or Soat2 confirmed the importance of their gene products on cholesterol esterification and lipoprotein physiology. Although these studies supported clinical trials which used non-selective ACAT inhibitors, these trials did not report benefits, and one showed an increased risk. Early genetic studies have implicated common variants in both genes with human traits, including lipoprotein levels, coronary artery disease, and Alzheimer’s disease; however, modern genome-wide association studies have not replicated these associations. In contrast, the common SOAT1 variants are most reproducibly associated with testosterone levels. Keywords: cholesterol esterification; atherosclerosis; ACAT; SOAT; inhibitors; clinical trial Citation: Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its 1. Introduction Discovery to Clinical Trials and the The acyl-coenzyme A:cholesterol acyltransferase (ACAT; EC 2.3.1.26) enzyme family Genomics Era. Metabolites 2021, 11, consists of membrane-spanning proteins, which are primarily located in the endoplasmic 543. https://doi.org/10.3390/ reticulum [1]. -

Phenotype Heterogeneity of Congenital Adrenal Hyperplasia Due
Kor et al. BMC Medical Genetics (2018) 19:115 https://doi.org/10.1186/s12881-018-0629-2 CASE REPORT Open Access Phenotype heterogeneity of congenital adrenal hyperplasia due to genetic mosaicism and concomitant nephrogenic diabetes insipidus in a sibling Yılmaz Kor1†, Minjing Zou2†, Roua A. Al-Rijjal2, Dorota Monies2, Brian F. Meyer2 and Yufei Shi2* Abstract Background: Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21OHD) is an autosomal recessive disorder caused by mutations in the CYP21A2. Congenital nephrogenic diabetes insipidus (NDI) is a rare X- linked recessive or autosomal recessive disorder caused by mutations in either AVPR2 or AQP2. Genotype-phenotype discordance caused by genetic mosaicism in CAH patients has not been reported, nor the concomitant CAH and NDI. Case presentation: We investigated a patient with concomitant CAH and NDI from a consanguineous family. She (S- 1) presented with clitoromegaly at 3 month of age, and polydipsia and polyuria at 13 month of age. Her parents and two elder sisters (S-2 and S-3) were clinically normal, but elevated levels of serum 17-hydroxyprogesterone (17-OHP) were observed in the mother and S-2. The coding region of CYP21A2 and AQP2 were analyzed by PCR-sequencing analysis to identify genetic defects. Two homozygous CYP21A2 mutations (p.R357W and p.P454S) were identified in the proband and her mother and S-2. The apparent genotype-phenotype discordance was due to presence of small amount of wild-type CYP21A2 alleles in S-1, S-2, and their mother’s genome, thus protecting them from development of classic form of 21OHD (C21OHD). -

Hsd17b1) Inhibitor for Endometriosis
DEVELOPMENT OF HYDROXYSTEROID (17-BETA) DEHYDROGENASE TYPE 1 (HSD17B1) INHIBITOR FOR ENDOMETRIOSIS Niina Saarinen1,2, Tero Linnanen1, Jasmin Tiala1, Camilla Stjernschantz1, Leena Hirvelä1, Taija Heinosalo2, Bert Delvoux3, Andrea Romano3, Gabriele Möller4, Jerzy Adamski4, Matti Poutanen2, Pasi Koskimies1 1Forendo Pharma Ltd, Finland; 2Institute of Biomedicine, Research Centre for Integrative Physiology and Pharmacology, University of Turku, Finland; 3Department of Obstetrics and Gynaecology; GROW, School for Oncology and Developmental Biology; Maastricht University Medical Centre, The Netherlands; 4Institute of Experimental Genetics, Genome Analysis Center, Helmholtz Zentrum München, Germany BACKGROUND OBJECTIVE Local activation of estrogens in endometriosis tissue The main objective of the present work was to assess is considered important for growth of the lesions. the preclinical efficacy of the novel HSD17B1 inhibitor, Hydroxysteroid (17-beta) dehydrogenase type 1 FOR-6219 (HSD17B1) is expressed in endometriosis tissue and converts the biologically low-active estrogen, estrone (E1), to the highly active estradiol (E2), while hydroxysteroid (17-beta) dehydrogenase type 2 (HSD17B2), catalyzes the opposite reaction. In contrast to eutopic endometrium, in endometriotic lesions the HSD17B1/HSD17B2 expression ratio is increased and E2 levels are higher than those of E1 throughout the menstrual cycle. Thus, inhibition of HSD17B1 is considered as a feasible strategy for lowering local E2 production in endometriosis. MAIN RESULTS FOR-6219 inhibits human HSD17B1 Ø FOR-6219 is a potent and FOR-6219 does not trigger estrogenic fully selective inhibitor of response in immature rat uterine human HSD17B1 over growth assay HSD17B2 Ø FOR-6219 does not bind to estrogen receptor α or β, and exhibits no estrogen-like response in immature rat uterotrophic assay Ø FOR-6219 inhibits HSD17B1 in cynomolgus monkey, dog and rabbit i.e. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Functional Silencing of HSD17B2 in Prostate Cancer Promotes Disease Progression
Published OnlineFirst September 18, 2018; DOI: 10.1158/1078-0432.CCR-18-2392 Translational Cancer Mechanisms and Therapy Clinical Cancer Research Functional Silencing of HSD17B2 in Prostate Cancer Promotes Disease Progression Xiaomei Gao1,2, Charles Dai3, Shengsong Huang4, Jingjie Tang1,2, Guoyuan Chen1, Jianneng Li3, Ziqi Zhu3, Xuyou Zhu5, Shuirong Zhou1,2, Yuanyuan Gao1,2, Zemin Hou1,2, Zijun Fang1,2, Chengdang Xu4, Jianyang Wang1,2, Denglong Wu4, Nima Sharifi3,6,7, and Zhenfei Li1,2 Abstract Purpose: Steroidogenic enzymes are essential for prostate (DHT) to each of their upstream precursors. HSD17B2 over- cancer development. Enzymes inactivating potent androgens expression suppressed androgen-induced cell proliferation were not investigated thoroughly, which leads to limited inter- and xenograft growth. Multiple mechanisms were involved ference strategies for prostate cancer therapy. Here we charac- in HSD17B2 functional silencing including DNA methylation terizedtheclinical relevance,significance, andregulation mech- and mRNA alternative splicing. DNA methylation decreased anism of enzyme HSD17B2 in prostate cancer development. the HSD17B2 mRNA level. Two new catalytic-deficient iso- Experimental Design: HSD17B2 expression was detected forms, generated by alternative splicing, bound to wild-type with patient specimens and prostate cancer cell lines. Function 17bHSD2 and promoted its degradation. Splicing factors of HSD17B2 in steroidogenesis, androgen receptor (AR) sig- SRSF1 and SRSF5 participated in the generation of new naling, and tumor growth was investigated with prostate isoforms. cancer cell lines and a xenograft model. DNA methylation Conclusions: Our findings provide evidence of the clinical and mRNA alternative splicing were investigated to unveil the relevance, significance, and regulation of HSD17B2 in prostate mechanisms of HSD17B2 regulation. -
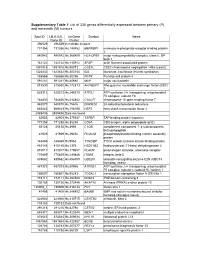
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Treating Endometriosis
ADVERTISEMENT FEATURE Forendo Pharma forendo.com Treating endometriosis By using a tissue-specific hormone inhibitor to rebalance local estrogen metabolism, Forendo Pharma could provide long-term treatment to millions of women suffering from endometriosis. With its expertise in tissue-specific hormone O OH therapies, the Finnish company Forendo Pharma HSD17B1 is tackling endometriosis, a condition that affects 10% of women of childbearing age. “Endometriosis is a difficult condition to treat, mainly because the estrogen-deficiency symptoms generated by HSD17B2 the currently used drugs prevent long-term use. HO HO profile Unlike these therapies, our strategy uses novel Estrone (E1) Estradiol (E2) 17-β-hydroxysteroid dehydrogenase (HSD17B) * Low activity * Highly active inhibitors which act locally, without impacting the Figure 1: Forendo’s FOR-6219. The basic concept behind the HSD17B1 inhibitor involves blockage of the systemic estrogen level,” said company CEO Risto conversion of estrone to estradiol. Lammintausta. The company was founded in 2013 by Finnish estrogen action, by converting non-active estrone cannot be controlled with hormonal therapies or drug development pioneers to exploit the find- into active estradiol within endometrial cells. When even surgery. “Whilst more efficient tools for diagno- ings of leading endocrinology researchers Matti this pathway is blocked, the build-up of high levels sis also need to be developed in order to provide an Poutanen and Antti Perheentupa, from the University of the estrogenic hormone estradiol is prevented, opportunity to treat women at an earlier stage and of Turku and Turku University Hospital, Finland. Led which will limit the ability of endometrial cells to form prevent these problems, HSD17B1 inhibitors offer a by Lammintausta, who has over 30 years of experi- endometriotic lesions. -

Evidence for Cross Species Extrapolation of Mammalian-Based High-Throughput Screening Assay Results † † ‡ † Carlie A
Article Cite This: Environ. Sci. Technol. 2018, 52, 13960−13971 pubs.acs.org/est Evidence for Cross Species Extrapolation of Mammalian-Based High-Throughput Screening Assay Results † † ‡ † Carlie A. LaLone,*, Daniel L. Villeneuve, Jon A. Doering, Brett R. Blackwell, § § ¶ † || Thomas R. Transue, Cody W. Simmons, Joe Swintek, Sigmund J. Degitz, Antony J. Williams, † and Gerald T. Ankley † Office of Research and Development, National Health and Environmental Effects Research Laboratory, Mid-Continent Ecology Division, US Environmental Protection Agency, 6201 Congdon Blvd., Duluth, Minnesota 55804, United States ‡ National Research Council, 6201 Congdon Blvd., Duluth, Minnesota 55804, United States § CSRA Inc., 109 T.W. Alexander Drive, Research Triangle Park, North Carolina 27711, United States ¶ Badger Technical Services, 6201 Congdon Blvd., Duluth, Minnesota 55804, United States || Office of Research and Development, National Center for Computational Toxicology, US Environmental Protection Agency, 109 T.W. Alexander Drive, Research Triangle Park, North Carolina 27711, United States *S Supporting Information ABSTRACT: High-throughput screening (HTS) and computational technologies have emerged as important tools for chemical hazard identification. The US Environmental Protection Agency (EPA) launched the Toxicity ForeCaster (ToxCast) Program, which has screened thousands of chemicals in hundreds of mammalian-based HTS assays for biological activity. The data are being used to prioritize toxicity testing on those chemi- cals likely to lead to adverse effects. To use HTS assays in predicting hazard to both humans and wildlife, it is necessary to understand how broadly these data may be extrapolated across species. The US EPA Sequence Alignment to Predict Across Species Susceptibility (SeqAPASS; https://seqapass.epa.gov/seqapass/) tool was used to assess conservation of the 484 protein targets represented in the suite of ToxCast assays and other HTS assays. -

WO 2018/190970 Al 18 October 2018 (18.10.2018) W !P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/190970 Al 18 October 2018 (18.10.2018) W !P O PCT (51) International Patent Classification: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, CI2Q 1/32 (2006.01) UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, (21) International Application Number: EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, PCT/US2018/021 109 MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, (22) International Filing Date: TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, 06 March 2018 (06.03.2018) KM, ML, MR, NE, SN, TD, TG). (25) Filing Language: English Declarations under Rule 4.17: (26) Publication Langi English — as to applicant's entitlement to apply for and be granted a patent (Rule 4.1 7(H)) (30) Priority Data: — as to the applicant's entitlement to claim the priority of the 62/484,141 11 April 2017 ( 11.04.2017) US earlier application (Rule 4.17(Hi)) (71) Applicant: REGENERON PHARMACEUTICALS, Published: INC. [US/US]; 777 Old Saw Mill River Road, Tarrytown, — with international search report (Art. 21(3)) New York 10591-6707 (US). — with sequence listing part of description (Rule 5.2(a)) (72) Inventors: STEVIS, Panayiotis; 777 Old Saw Mill Riv er Road, Tarrytown, New York 10591-6707 (US). -

Effects on Steroid 5-Alpha Reductase Gene Expression of Thai Rice Bran Extracts and Molecular Dynamics Study on SRD5A2
biology Article Effects on Steroid 5-Alpha Reductase Gene Expression of Thai Rice Bran Extracts and Molecular Dynamics Study on SRD5A2 Chiranan Khantham 1 , Wipawadee Yooin 1,2 , Korawan Sringarm 2,3 , Sarana Rose Sommano 2,4 , Supat Jiranusornkul 1, Francisco David Carmona 5,6 , Wutigri Nimlamool 7 , Pensak Jantrawut 1,2, Pornchai Rachtanapun 8,9 and Warintorn Ruksiriwanich 1,2,8,* 1 Department of Pharmaceutical Sciences, Faculty of Pharmacy, Chiang Mai University, Chiang Mai 50200, Thailand; [email protected] (C.K.); [email protected] (W.Y.); [email protected] (S.J.); [email protected] (P.J.) 2 Cluster of Research and Development of Pharmaceutical and Natural Products Innovation for Human or Animal, Chiang Mai University, Chiang Mai 50200, Thailand; [email protected] (K.S.); [email protected] (S.R.S.) 3 Department of Animal and Aquatic Sciences, Faculty of Agriculture, Chiang Mai University, Chiang Mai 50200, Thailand 4 Plant Bioactive Compound Laboratory (BAC), Department of Plant and Soil Sciences, Faculty of Agriculture, Chiang Mai University, Chiang Mai 50200, Thailand 5 Departamento de Genética e Instituto de Biotecnología, Universidad de Granada, 18071 Granada, Spain; [email protected] 6 Instituto de Investigación Biosanitaria ibs.GRANADA, 18014 Granada, Spain 7 Citation: Khantham, C.; Yooin, W.; Department of Pharmacology, Faculty of Medicine, Chiang Mai University, Chiang Mai 50200, Thailand; Sringarm, K.; Sommano, S.R.; [email protected] 8 Cluster of Agro Bio-Circular-Green Industry, Faculty of Agro-Industry, Chiang Mai University, Jiranusornkul, S.; Carmona, F.D.; Chiang Mai 50100, Thailand; [email protected] Nimlamool, W.; Jantrawut, P.; 9 School of Agro-Industry, Faculty of Agro-Industry, Chiang Mai University, Chiang Mai 50100, Thailand Rachtanapun, P.; Ruksiriwanich, W. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota.