The Neurodegenerative Diseases ALS and SMA Are Linked at The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Identification of the Causative Gene for Simmental Arachnomelia Syndrome Using a Network-Based Disease Gene Prioritization Approach
Identification of the Causative Gene for Simmental Arachnomelia Syndrome Using a Network-Based Disease Gene Prioritization Approach Shihui Jiao1., Qin Chu2., Yachun Wang1*, Zhenquan Xie3, Shiyu Hou3, Airong Liu5, Hongjun Wu4, Lin Liu6, Fanjun Geng7, Congyong Wang7, Chunhua Qin8, Rui Tan9, Xixia Huang10, Shixin Tan11, Meng Wu12, Xianzhou Xu12, Xuan Liu1, Ying Yu1, Yuan Zhang1 1 Key Laboratory of Agricultural Animal and Breeding, National Engineering Laboratory for Animal Breeding, College of Animal Science and Technology, China Agricultural University, Beijing, China, 2 Institute of Animal Husbandry and Veterinary Medicine, Beijing Academy of Agriculture and Forestry Sciences, Beijing, China, 3 Anshan Hengli Dairy Farm, Anshan, Liaoning, China, 4 Xiertala Breeding Farm, Hailaer Farm Buro, Hailaer, Inner Mongolia, China, 5 Hailaer Farm Buro, Hailaer, Inner Mongolia, China, 6 Beijing Dairy Cattle Centre, Beijing, China, 7 Dingyuan Seedstock Bulls Breeding Ltd. Company, Zhengzhou, Henan, China, 8 Ningxia Sygen BioEngineering Research Center, Yinchuan, Ningxia, China, 9 Xinjiang General Livestock Service, Urumqi, Xinjiang, China, 10 College of Animal Science, Xinjiang Agriculture University, Urumqi, Xinjiang, China, 11 Xinjiang Tianshan Animal Husbandry Bio-engineering Co. Ltd, Urumqi, Xinjiang, China, 12 Dalian Xuelong Industry Limited Group, Dalian, Liaoning, China Abstract Arachnomelia syndrome (AS), mainly found in Brown Swiss and Simmental cattle, is a congenital lethal genetic malformation of the skeletal system. In this study, a network-based disease gene prioritization approach was implemented to rank genes in the previously reported ,7 Mb region on chromosome 23 associated with AS in Simmental cattle. The top 6 ranked candidate genes were sequenced in four German Simmental bulls, one known AS-carrier ROMEL and a pooled sample of three known non-carriers (BOSSAG, RIFURT and HIRMER). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Hypopituitarism May Be an Additional Feature of SIM1 and POU3F2 Containing 6Q16 Deletions in Children with Early Onset Obesity
Open Access Journal of Pediatric Endocrinology Case Report Hypopituitarism may be an Additional Feature of SIM1 and POU3F2 Containing 6q16 Deletions in Children with Early Onset Obesity Rutteman B1, De Rademaeker M2, Gies I1, Van den Bogaert A2, Zeevaert R1, Vanbesien J1 and De Abstract Schepper J1* Over the past decades, 6q16 deletions have become recognized as a 1Department of Pediatric Endocrinology, UZ Brussel, frequent cause of the Prader-Willi-like syndrome. Involvement of SIM1 in the Belgium deletion has been linked to the development of obesity. Although SIM1 together 2Department of Genetics, UZ Brussel, Belgium with POU3F2, which is located close to the SIM1 locus, are involved in pituitary *Corresponding author: De Schepper J, Department development and function, pituitary dysfunction has not been reported frequently of Pediatric Endocrinology, UZ Brussel, Belgium in cases of 6q16.1q16.3 deletion involving both genes. Here we report on a case of a girl with typical Prader-Willi-like symptoms including early-onset Received: December 23, 2016; Accepted: February 21, hyperphagic obesity, hypotonia, short hands and feet and neuro-psychomotor 2017; Published: February 22, 2017 development delay. Furthermore, she suffered from central diabetes insipidus, central hypothyroidism and hypocortisolism. Her genetic defect is a 6q16.1q16.3 deletion, including SIM1 and POU3F2. We recommend searching for a 6q16 deletion in children with early onset hyperphagic obesity associated with biological signs of hypopituitarism and/or polyuria-polydipsia syndrome. The finding of a combined SIM1 and POU3F2 deletion should prompt monitoring for the development of hypopituitarism, if not already present at diagnosis. Keywords: 6q16 deletion; SIM1; POU3F2; Prader-Willi-like syndrome; Hypopituitarism Introduction insipidus and of some specific pituitary hormone deficiencies in children with a combined SIM1 and POU3F2 deficiency. -

Whole Exome Sequencing in ADHD Trios from Single and Multi-Incident Families Implicates New Candidate Genes and Highlights Polygenic Transmission
European Journal of Human Genetics (2020) 28:1098–1110 https://doi.org/10.1038/s41431-020-0619-7 ARTICLE Whole exome sequencing in ADHD trios from single and multi-incident families implicates new candidate genes and highlights polygenic transmission 1,2 1 1,2 1,3 1 Bashayer R. Al-Mubarak ● Aisha Omar ● Batoul Baz ● Basma Al-Abdulaziz ● Amna I. Magrashi ● 1,2 2 2,4 2,4,5 6 Eman Al-Yemni ● Amjad Jabaan ● Dorota Monies ● Mohamed Abouelhoda ● Dejene Abebe ● 7 1,2 Mohammad Ghaziuddin ● Nada A. Al-Tassan Received: 26 September 2019 / Revised: 26 February 2020 / Accepted: 10 March 2020 / Published online: 1 April 2020 © The Author(s) 2020. This article is published with open access Abstract Several types of genetic alterations occurring at numerous loci have been described in attention deficit hyperactivity disorder (ADHD). However, the role of rare single nucleotide variants (SNVs) remains under investigated. Here, we sought to identify rare SNVs with predicted deleterious effect that may contribute to ADHD risk. We chose to study ADHD families (including multi-incident) from a population with a high rate of consanguinity in which genetic risk factors tend to 1234567890();,: 1234567890();,: accumulate and therefore increasing the chance of detecting risk alleles. We employed whole exome sequencing (WES) to interrogate the entire coding region of 16 trios with ADHD. We also performed enrichment analysis on our final list of genes to identify the overrepresented biological processes. A total of 32 rare variants with predicted damaging effect were identified in 31 genes. At least two variants were detected per proband, most of which were not exclusive to the affected individuals. -

Genome-Wide Association Study for Circulating Fibroblast Growth Factor
www.nature.com/scientificreports OPEN Genome‑wide association study for circulating fbroblast growth factor 21 and 23 Gwo‑Tsann Chuang1,2,14, Pi‑Hua Liu3,4,14, Tsui‑Wei Chyan5, Chen‑Hao Huang5, Yu‑Yao Huang4,6, Chia‑Hung Lin4,7, Jou‑Wei Lin8, Chih‑Neng Hsu8, Ru‑Yi Tsai8, Meng‑Lun Hsieh9, Hsiao‑Lin Lee9, Wei‑shun Yang2,9, Cassianne Robinson‑Cohen10, Chia‑Ni Hsiung11, Chen‑Yang Shen12,13 & Yi‑Cheng Chang2,9,12* Fibroblast growth factors (FGFs) 21 and 23 are recently identifed hormones regulating metabolism of glucose, lipid, phosphate and vitamin D. Here we conducted a genome‑wide association study (GWAS) for circulating FGF21 and FGF23 concentrations to identify their genetic determinants. We enrolled 5,000 participants from Taiwan Biobank for this GWAS. After excluding participants with diabetes mellitus and quality control, association of single nucleotide polymorphisms (SNPs) with log‑transformed FGF21 and FGF23 serum concentrations adjusted for age, sex and principal components of ancestry were analyzed. A second model additionally adjusted for body mass index (BMI) and a third model additionally adjusted for BMI and estimated glomerular fltration rate (eGFR) were used. A total of 4,201 participants underwent GWAS analysis. rs67327215, located within RGS6 (a gene involved in fatty acid synthesis), and two other SNPs (rs12565114 and rs9520257, located between PHC2-ZSCAN20 and ARGLU1-FAM155A respectively) showed suggestive associations with serum FGF21 level (P = 6.66 × 10–7, 6.00 × 10–7 and 6.11 × 10–7 respectively). The SNPs rs17111495 and rs17843626 were signifcantly associated with FGF23 level, with the former near PCSK9 gene and the latter near HLA-DQA1 gene (P = 1.04 × 10–10 and 1.80 × 10–8 respectively). -

Controls Homeostatic Splicing of ARGLU1 Mrna Stephan P
Published online 28 November 2016 Nucleic Acids Research, 2017, Vol. 45, No. 6 3473–3486 doi: 10.1093/nar/gkw1140 An Ultraconserved Element (UCE) controls homeostatic splicing of ARGLU1 mRNA Stephan P. Pirnie, Ahmad Osman, Yinzhou Zhu and Gordon G. Carmichael* Department of Genetics and Genome Sciences, UCONN Health Center, 400 Farmington Avenue, Farmington, CT 06030, USA Received January 13, 2016; Revised October 25, 2016; Editorial Decision October 28, 2016; Accepted October 31, 2016 ABSTRACT or inhibit the usage of a particular splice site (1,2). The ex- pression of trans-acting factors in a developmental and tis- Arginine and Glutamate-Rich protein 1 (ARGLU1) is sue specific manner results in regulated splicing that isof- a protein whose function is poorly understood, but ten cell specific. Most notably, trans-acting proteins such as may act in both transcription and pre-mRNA splicing. the NOVA (3–6), the RBFOX (7) and SR protein (8–11) We demonstrate that the ARGLU1 gene expresses families, and a number of hnRNP (12,13) proteins compete at least three distinct RNA splice isoforms – a fully to bind nascent RNAs at specific motifs and drive regula- spliced isoform coding for the protein, an isoform tion of alternative splicing in a tissue and developmentally containing a retained intron that is detained in the specific manner. Alternative splicing is an important pro- nucleus, and an isoform containing an alternative cess that is seen in at least 95% of multi-exon genes in the exon that targets the transcript for nonsense medi- human transcriptome (14). Furthermore, alternative splic- ated decay. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality. -

Mutational Landscape Differences Between Young-Onset and Older-Onset Breast Cancer Patients Nicole E
Mealey et al. BMC Cancer (2020) 20:212 https://doi.org/10.1186/s12885-020-6684-z RESEARCH ARTICLE Open Access Mutational landscape differences between young-onset and older-onset breast cancer patients Nicole E. Mealey1 , Dylan E. O’Sullivan2 , Joy Pader3 , Yibing Ruan3 , Edwin Wang4 , May Lynn Quan1,5,6 and Darren R. Brenner1,3,5* Abstract Background: The incidence of breast cancer among young women (aged ≤40 years) has increased in North America and Europe. Fewer than 10% of cases among young women are attributable to inherited BRCA1 or BRCA2 mutations, suggesting an important role for somatic mutations. This study investigated genomic differences between young- and older-onset breast tumours. Methods: In this study we characterized the mutational landscape of 89 young-onset breast tumours (≤40 years) and examined differences with 949 older-onset tumours (> 40 years) using data from The Cancer Genome Atlas. We examined mutated genes, mutational load, and types of mutations. We used complementary R packages “deconstructSigs” and “SomaticSignatures” to extract mutational signatures. A recursively partitioned mixture model was used to identify whether combinations of mutational signatures were related to age of onset. Results: Older patients had a higher proportion of mutations in PIK3CA, CDH1, and MAP3K1 genes, while young- onset patients had a higher proportion of mutations in GATA3 and CTNNB1. Mutational load was lower for young- onset tumours, and a higher proportion of these mutations were C > A mutations, but a lower proportion were C > T mutations compared to older-onset tumours. The most common mutational signatures identified in both age groups were signatures 1 and 3 from the COSMIC database. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Investigating the Molecular Mechanisms of Splicing-Perturbing Small Molecules with Massively Parallel Sequencing in a Myotonic Dystrophy 1 Model
INVESTIGATING THE MOLECULAR MECHANISMS OF SPLICING-PERTURBING SMALL MOLECULES WITH MASSIVELY PARALLEL SEQUENCING IN A MYOTONIC DYSTROPHY 1 MODEL by MATTHEW TANNER A THESIS Presented to the Department of Chemistry and Biochemistry and the Robert D. Clark Honors College in partial fulfillment of the requirements for the degree of Bachelor of Science June, 2014 An Abstract of the Thesis of Matthew Tanner for the degree of Bachelor of Science in the Department of Chemistry and Biochemistry to be taken June, 2014 Title: Investigating the Molecular Mechanisms of Splicing-Perturbing Small Molecules with Massively Parallel Sequencing in a Myotonic Dystrophy 1 Model Approved: 4 fhh 73cJ-i J. Andrew Berglund Myotonic dystrophy is the most common form of adult-onset muscular dystrophy and appears in two forms: myotonic dystrophy 1 (OM 1) and 2 (DM2). Both diseases arc characterized by progressive muscle degeneration, myotonia. iridescent cataracts, and in severe cases neurodcgcncration and cardiac dysfunction. Both fonns of myotonic dystrophy arc caused by an expansion of repeat DNA in distinct loci in the genome. In DM 1, a CTG repeat is expanded from less than 50 repeats in nonnal individuals to up to several thousand repeats in DM 1 patients. The molecular basis of this disease relics on the transcription of these repeats from DNA into RNA. Small molecules that can specifically target the repeats at the DNA level and inhibit their transcription - and thus alleviate the disease symptoms - represent a prime target for the development of therapeutics. This study investigates the capacity of two small molecules, pentamidine and actinomycin D, to reverse the molecular symptoms of DM I through transcriptional inhibition and provides insight into their DNA target specificity and potential mechanisms of action. -
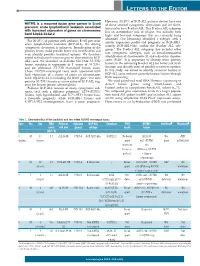
NUTM1 Is a Recurrent Fusion Gene Partner in B-Cell Precursor Acute
LETTERS TO THE EDITOR However, 20-25% of BCP-ALL patients do not have one NUTM1 is a recurrent fusion gene partner in B-cell of these sentinel cytogenetic aberrations and are there- precursor acute lymphoblastic leukemia associated fore said to have B-other ALL. This B-other ALL subgroup with increased expression of genes on chromosome has an intermediate risk of relapse, but includes both band 10p12.31-12.2 high- and low-risk subgroups that are currently being identified. Our laboratory identified a subtype with a For 20-25% of patients with pediatric B-cell precursor similar expression profile and prognosis as BCR-ABL1, acute lymphoblastic leukemia (BCP-ALL), the driving namely BCR-ABL1-like, within the B-other ALL sub- cytogenetic aberration is unknown. Identification of the group.2 The B-other ALL subgroup also includes other primary lesion could provide better risk stratification and rare cytogenetic subtypes, such as intrachromosomal even identify possible treatment options. We therefore amplification of chromosome 21 and a dicentric chromo- aimed to find novel recurrent genetic aberrations in BCP- 1 ALL cases. We identified an in-frame SLC12A6-NUTM1 some (9;20). It is important to identify more primary fusion, resulting in expression of 3’ exons of NUTM1, lesions in the remaining B-other ALL for better risk strat- and six additional NUTM1-rearranged fusion cases. ification and identification of possible treatment options. These NUTM1-rearranged cases were associated with In this study, we aimed to identify recurrent fusions in high expression of a cluster of genes on chromosome BCP-ALL cases without currently known lesions through band 10p12.31-12.2, including the BMI1 gene. -

CCAR1 Polyclonal Antibody
PRODUCT DATA SHEET Bioworld Technology,Inc. CCAR1 polyclonal antibody Catalog: BS8140 Host: Rabbit Reactivity: Human BackGround: by affinity-chromatography using epitope-specific im- CARP-1 (cell division cycle and apoptosis regulator 1), munogen and the purity is > 95% (by SDS-PAGE). also known as CCAR1 or DIS, is a 1,150 amino acid pro- Applications: tein that localizes to the perinuclear region of the cyto- WB 1:500 - 1:2000 plasm and contains one SAP domain. Expressed in sever- IF 1:50 - 1:200 al epithelial cancer cell lines, including breast, colon, IP 1:50 - 1:100 prostate and leukemia, CARP-1 is involved in apoptotic Storage&Stability: signaling, as well as in cell cycle progression and cell Store at 4°C short term. Aliquot and store at -20°C long proliferation via interaction with c-Myc and cyclin B1. term. Avoid freeze-thaw cycles. CARP-1 is subject to DNA damage-induced phosphory- Specificity: lation, probably by ATM or ATR. The gene encoding CCAR1 polyclonal antibody detects endogenous levels of CARP-1 maps to human chromosome 10, which houses CCAR1 protein. over 1,200 genes and comprises nearly 4.5% of the hu- DATA: man genome. Defects in some of the genes that map to chromosome 10 are associated with Charcot-Marie Tooth disease, Jackson-Weiss syndrome, Usher syndrome, non- syndromatic deafness, Wolman’s syndrome, Cowden syndrome, multiple endocrine neoplasia type 2 and por- phyria. Product: Rabbit IgG, 1mg/ml in PBS with 0.02% sodium azide, WesternBlot (WB) analysis of CCAR1 polyclonal antibody 50% glycerol, pH7.2 Note: Molecular Weight: For research use only, not for use in diagnostic procedure.