Bioinformatics to Integrate Protein and Gene Information in a Relational Context, Application to Human Proteomic and Transcriptomic Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Astrin-SKAP Complex Reconstitution Reveals Its Kinetochore
RESEARCH ARTICLE Astrin-SKAP complex reconstitution reveals its kinetochore interaction with microtubule-bound Ndc80 David M Kern1,2, Julie K Monda1,2†, Kuan-Chung Su1†, Elizabeth M Wilson-Kubalek3, Iain M Cheeseman1,2* 1Whitehead Institute for Biomedical Research, Cambridge, United States; 2Department of Biology, Massachusetts Institute of Technology, Cambridge, United States; 3Department of Cell Biology, The Scripps Research Institute, La Jolla, United States Abstract Chromosome segregation requires robust interactions between the macromolecular kinetochore structure and dynamic microtubule polymers. A key outstanding question is how kinetochore-microtubule attachments are modulated to ensure that bi-oriented attachments are selectively stabilized and maintained. The Astrin-SKAP complex localizes preferentially to properly bi-oriented sister kinetochores, representing the final outer kinetochore component recruited prior to anaphase onset. Here, we reconstitute the 4-subunit Astrin-SKAP complex, including a novel MYCBP subunit. Our work demonstrates that the Astrin-SKAP complex contains separable kinetochore localization and microtubule binding domains. In addition, through cross-linking analysis in human cells and biochemical reconstitution, we show that the Astrin-SKAP complex binds synergistically to microtubules with the Ndc80 complex to form an integrated interface. We propose a model in which the Astrin-SKAP complex acts together with the Ndc80 complex to stabilize correctly formed kinetochore-microtubule interactions. *For correspondence: DOI: https://doi.org/10.7554/eLife.26866.001 [email protected] †These authors contributed equally to this work Introduction Competing interests: The The macromolecular kinetochore complex links chromosomes to dynamic microtubule polymers and authors declare that no harnesses the forces generated by microtubule growth and depolymerization to facilitate accurate competing interests exist. -

Aberrant Methylation Underlies Insulin Gene Expression in Human Insulinoma
ARTICLE https://doi.org/10.1038/s41467-020-18839-1 OPEN Aberrant methylation underlies insulin gene expression in human insulinoma Esra Karakose1,6, Huan Wang 2,6, William Inabnet1, Rajesh V. Thakker 3, Steven Libutti4, Gustavo Fernandez-Ranvier 1, Hyunsuk Suh1, Mark Stevenson 3, Yayoi Kinoshita1, Michael Donovan1, Yevgeniy Antipin1,2, Yan Li5, Xiaoxiao Liu 5, Fulai Jin 5, Peng Wang 1, Andrew Uzilov 1,2, ✉ Carmen Argmann 1, Eric E. Schadt 1,2, Andrew F. Stewart 1,7 , Donald K. Scott 1,7 & Luca Lambertini 1,6 1234567890():,; Human insulinomas are rare, benign, slowly proliferating, insulin-producing beta cell tumors that provide a molecular “recipe” or “roadmap” for pathways that control human beta cell regeneration. An earlier study revealed abnormal methylation in the imprinted p15.5-p15.4 region of chromosome 11, known to be abnormally methylated in another disorder of expanded beta cell mass and function: the focal variant of congenital hyperinsulinism. Here, we compare deep DNA methylome sequencing on 19 human insulinomas, and five sets of normal beta cells. We find a remarkably consistent, abnormal methylation pattern in insu- linomas. The findings suggest that abnormal insulin (INS) promoter methylation and altered transcription factor expression create alternative drivers of INS expression, replacing cano- nical PDX1-driven beta cell specification with a pathological, looping, distal enhancer-based form of transcriptional regulation. Finally, NFaT transcription factors, rather than the cano- nical PDX1 enhancer complex, are predicted to drive INS transactivation. 1 From the Diabetes Obesity and Metabolism Institute, The Department of Surgery, The Department of Pathology, The Department of Genetics and Genomics Sciences and The Institute for Genomics and Multiscale Biology, The Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA. -
Primepcr™Assay Validation Report
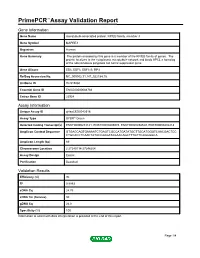
PrimePCR™Assay Validation Report Gene Information Gene Name microtubule-associated protein, RP/EB family, member 3 Gene Symbol MAPRE3 Organism Human Gene Summary The protein encoded by this gene is a member of the RP/EB family of genes. The protein localizes to the cytoplasmic microtubule network and binds APCL a homolog of the adenomatous polyposis coli tumor suppressor gene. Gene Aliases EB3, EBF3, EBF3-S, RP3 RefSeq Accession No. NC_000002.11, NT_022184.15 UniGene ID Hs.515860 Ensembl Gene ID ENSG00000084764 Entrez Gene ID 22924 Assay Information Unique Assay ID qHsaCED0042518 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233121, ENST00000405074, ENST00000458529, ENST00000402218 Amplicon Context Sequence GTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGGTCAACGACTCC CTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCA Amplicon Length (bp) 69 Chromosome Location 2:27245114-27246204 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 90 R2 0.9993 cDNA Cq 24.76 cDNA Tm (Celsius) 80 gDNA Cq 26.8 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report MAPRE3, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

Epigenetic Regulation of the Human Genome by Transposable Elements
EPIGENETIC REGULATION OF THE HUMAN GENOME BY TRANSPOSABLE ELEMENTS A Dissertation Presented to The Academic Faculty By Ahsan Huda In Partial Fulfillment Of the Requirements for the Degree Doctor of Philosophy in Bioinformatics in the School of Biology Georgia Institute of Technology August 2010 EPIGENETIC REGULATION OF THE HUMAN GENOME BY TRANSPOSABLE ELEMENTS Approved by: Dr. I. King Jordan, Advisor Dr. John F. McDonald School of Biology School of Biology Georgia Institute of Technology Georgia Institute of Technology Dr. Leonardo Mariño-Ramírez Dr. Jung Choi NCBI/NLM/NIH School of Biology Georgia Institute of Technology Dr. Soojin Yi, School of Biology Georgia Institute of Technology Date Approved: June 25, 2010 To my mother, your life is my inspiration... ACKNOWLEDGEMENTS I am evermore thankful to my advisor Dr. I. King Jordan for his guidance, support and encouragement throughout my years as a PhD student. I am very fortunate to have him as my mentor as he is instrumental in shaping my personal and professional development. His contributions will continue to impact my life and career and for that I am forever grateful. I am also thankful to my committee members, John McDonald, Leonardo Mariño- Ramírez, Soojin Yi and Jung Choi for their continued support during my PhD career. Through my meetings and discussions with them, I have developed an appreciation for the scientific method and a thorough understanding of my field of study. I am especially grateful to my friends and colleagues, Lee Katz and Jittima Piriyapongsa for their support and presence, which brightened the atmosphere in the lab in the months and years past. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

Whole Exome Sequencing in ADHD Trios from Single and Multi-Incident Families Implicates New Candidate Genes and Highlights Polygenic Transmission
European Journal of Human Genetics (2020) 28:1098–1110 https://doi.org/10.1038/s41431-020-0619-7 ARTICLE Whole exome sequencing in ADHD trios from single and multi-incident families implicates new candidate genes and highlights polygenic transmission 1,2 1 1,2 1,3 1 Bashayer R. Al-Mubarak ● Aisha Omar ● Batoul Baz ● Basma Al-Abdulaziz ● Amna I. Magrashi ● 1,2 2 2,4 2,4,5 6 Eman Al-Yemni ● Amjad Jabaan ● Dorota Monies ● Mohamed Abouelhoda ● Dejene Abebe ● 7 1,2 Mohammad Ghaziuddin ● Nada A. Al-Tassan Received: 26 September 2019 / Revised: 26 February 2020 / Accepted: 10 March 2020 / Published online: 1 April 2020 © The Author(s) 2020. This article is published with open access Abstract Several types of genetic alterations occurring at numerous loci have been described in attention deficit hyperactivity disorder (ADHD). However, the role of rare single nucleotide variants (SNVs) remains under investigated. Here, we sought to identify rare SNVs with predicted deleterious effect that may contribute to ADHD risk. We chose to study ADHD families (including multi-incident) from a population with a high rate of consanguinity in which genetic risk factors tend to 1234567890();,: 1234567890();,: accumulate and therefore increasing the chance of detecting risk alleles. We employed whole exome sequencing (WES) to interrogate the entire coding region of 16 trios with ADHD. We also performed enrichment analysis on our final list of genes to identify the overrepresented biological processes. A total of 32 rare variants with predicted damaging effect were identified in 31 genes. At least two variants were detected per proband, most of which were not exclusive to the affected individuals. -

A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules Natasha L
www.nature.com/scientificreports OPEN A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules Natasha L. Patel-Murray1, Miriam Adam2, Nhan Huynh2, Brook T. Wassie2, Pamela Milani2 & Ernest Fraenkel 2,3* High-throughput screening and gene signature analyses frequently identify lead therapeutic compounds with unknown modes of action (MoAs), and the resulting uncertainties can lead to the failure of clinical trials. We developed an approach for uncovering MoAs through an interpretable machine learning model of transcriptomics, epigenomics, metabolomics, and proteomics. Examining compounds with benefcial efects in models of Huntington’s Disease, we found common MoAs for compounds with unrelated structures, connectivity scores, and binding targets. The approach also predicted highly divergent MoAs for two FDA-approved antihistamines. We experimentally validated these efects, demonstrating that one antihistamine activates autophagy, while the other targets bioenergetics. The use of multiple omics was essential, as some MoAs were virtually undetectable in specifc assays. Our approach does not require reference compounds or large databases of experimental data in related systems and thus can be applied to the study of agents with uncharacterized MoAs and to rare or understudied diseases. Unknown modes of action of drug candidates can lead to unpredicted consequences on efectiveness and safety. Computational methods, such as the analysis of gene signatures, and high-throughput experimental methods have accelerated the discovery of lead compounds that afect a specifc target or phenotype1–3. However, these advances have not dramatically changed the rate of drug approvals. Between 2000 and 2015, 86% of drug can- didates failed to earn FDA approval, with toxicity or a lack of efcacy being common reasons for their clinical trial termination4,5. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Produktinformation
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic MYCBP monoclonal antibody (M13), clone 1B12 Catalog # : H00026292-M13 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Transfected lysate) Description: MYCBP. Immunogen: MYCBP (NP_036465.2, 34 a.a. ~ 103 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: LYEEPEKPNSALDFLKHHLGAATPENPEIELLRLELAEMKEKYEAIVEENK KLKAKLAQYEPPQEEKRAE enlarge Host: Mouse Western Blot (Recombinant protein) Reactivity: Human Sandwich ELISA (Recombinant Isotype: IgG2a Kappa protein) Quality Control Antibody Reactive Against Recombinant Protein. Testing: enlarge ELISA Western Blot detection against Immunogen (33.44 KDa) . Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download Datasheet: Download Applications Western Blot (Transfected lysate) Page 1 of 3 2016/5/23 Western Blot analysis of MYCBP expression in transfected 293T cell line by MYCBP monoclonal antibody (M13), clone 1B12. Lane 1: MYCBP transfected lysate(12 KDa). Lane 2: Non-transfected lysate. Protocol Download Western Blot (Recombinant protein) Protocol Download Sandwich ELISA (Recombinant protein) Detection limit for recombinant GST tagged MYCBP is 0.1 ng/ml as a capture antibody. -

MAML1 Antibody A
Revision 1 C 0 2 - t MAML1 Antibody a e r o t S Orders: 877-616-CELL (2355) [email protected] Support: 877-678-TECH (8324) 8 0 Web: [email protected] 6 www.cellsignal.com 4 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source: UniProt ID: Entrez-Gene Id: WB, IP H Endogenous 130 Rabbit Q92585 9794 Product Usage Information Application Dilution Western Blotting 1:1000 Immunoprecipitation 1:50 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA and 50% glycerol. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity MAML1 Antibody detects endogenous levels of total MAML1 protein. It does not cross- react with MAML2 and MAML3. Species Reactivity: Human Source / Purification Polyclonal antibodies are produced by immunizing animals with a synthetic peptide corresponding to residues surrounding His811 of human MAML1. Antibodies are purified by protein A and peptide affinity chromatography. Background Mastermind-like (MAML) family of proteins are homologs of Drosophila Mastermind. The family is composed of three members in mammals: MAML1, MAML2, and MAML3 (1,2). MAML proteins form complexes with the intracellular domain of Notch (ICN) and the transcription factor CSL (RBP-Jκ) to regulate Notch target gene expression (3-5). MAML1 also interacts with myocyte enhancer factor 2C (MEF2C) to regulate myogenesis (6). MAML2 is frequently found to be fused with Mucoepidermoid carcinoma translocated gene 1 (MECT1, also know as WAMTP1 or TORC1) in patients with mucoepidermoid carcinomas and Warthin's tumors (7).