Direct Derivation of Benmoreite to Phonolite
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Source to Surface Model of Monogenetic Volcanism: a Critical Review
Downloaded from http://sp.lyellcollection.org/ by guest on September 28, 2021 Source to surface model of monogenetic volcanism: a critical review I. E. M. SMITH1 &K.NE´ METH2* 1School of Environment, University of Auckland, Auckland, New Zealand 2Volcanic Risk Solutions, Massey University, Palmerston North 4442, New Zealand *Correspondence: [email protected] Abstract: Small-scale volcanic systems are the most widespread type of volcanism on Earth and occur in all of the main tectonic settings. Most commonly, these systems erupt basaltic magmas within a wide compositional range from strongly silica undersaturated to saturated and oversatu- rated; less commonly, the spectrum includes more siliceous compositions. Small-scale volcanic systems are commonly monogenetic in the sense that they are represented at the Earth’s surface by fields of small volcanoes, each the product of a temporally restricted eruption of a composition- ally distinct batch of magma, and this is in contrast to polygenetic systems characterized by rela- tively large edifices built by multiple eruptions over longer periods of time involving magmas with diverse origins. Eruption styles of small-scale volcanoes range from pyroclastic to effusive, and are strongly controlled by the relative influence of the characteristics of the magmatic system and the surface environment. Gold Open Access: This article is published under the terms of the CC-BY 3.0 license. Small-scale basaltic magmatic systems characteris- hazards associated with eruptions, and this is tically occur at the Earth’s surface as fields of small particularly true where volcanic fields are in close monogenetic volcanoes. These volcanoes are the proximity to population centres. -

31 a Preliminary Study Of
31 A PRELIMINARY STUDY OF THE: TERTIARY VOLCANIC AND SEDIMENTARY ROCKS, GÜMELE, ESKİŞEHİR Eskişehir, Gümele Çevresindeki Tersiyer Volkanik ve Sedimanter Kayaçlarda Bir Ön Çalışma Taylan Lünel Middle East Technical University, Department of Geological Engineering Ankara ÖZ. — Seyitgazi-Eskişehir antiklinoriumu'nun çok fazla deforme olmuş ve metamorfizmaya uğramış kayaçlarının kuzey-kuzeybatısında bulunan sedimanter ve volkanik kayaçlar incelenmiştir. Karasal ve gölsel fasiyesde meydana gelen Tersi- yer sedimanter kayaçlar Güney Eskişehir küvetinde olunmuşlardır. Karasal fasiye- si meydana getiren kayaç birimlerini kaba kumtaşları, kumtaşları, bitki kalıntıları ihtiva eden kil ve marnlar ve serpantinit blokları taşıyan bazal konglomerası teşkil etmektedir. Gölsel fasiyes ise genellikle killi ve tüflü kalkerler, kalkerler, marnlar, kon- glomeralar ve tüflerden meydana gelmiştir. Küvetteki en eski sedimanlar ve piroklas- tikler Alt Miosen'de oluşmuşlardır. Yataya yakın konumlanmış bazik-intermediyar lav akıntıları Pliosen yaşlı olup Altüst Neojen sedimantasyon kesikliğinde meyda- na gelmiştir. Üst Neojen sedimanter kayaçları intermediyar-basaltik volkaniklerin üzerinde ince bandlar şeklinde bulunurlar. Bu birim marn ve kalkerlerden meyda- na gelmiştir. Alt Miosen’de asid volkanik faaliyetler neticesinde meydana gelen sil- lar (unweldd tuffs) oligomikt konglomeralardan evvel teşekkül etmiştir. Bu volkanik aktitivite muhtemelen kesikli ve kısıtlı olarak devam etmiş ve tüflü kalkerleri mey- dana getirmiştir. Pliosen yaşlı bazik-intermediyar -

THE HAWAIIAN-EMPEROR VOLCANIC CHAIN Part I Geologic Evolution
VOLCANISM IN HAWAII Chapter 1 - .-............,. THE HAWAIIAN-EMPEROR VOLCANIC CHAIN Part I Geologic Evolution By David A. Clague and G. Brent Dalrymple ABSTRACT chain, the near-fixity of the hot spot, the chemistry and timing of The Hawaiian-Emperor volcanic chain stretches nearly the eruptions from individual volcanoes, and the detailed geom 6,000 km across the North Pacific Ocean and consists of at least etry of volcanism. None of the geophysical hypotheses pro t 07 individual volcanoes with a total volume of about 1 million posed to date are fully satisfactory. However, the existence of km3• The chain is age progressive with still-active volcanoes at the Hawaiian ewell suggests that hot spots are indeed hot. In the southeast end and 80-75-Ma volcanoes at the northwest addition, both geophysical and geochemical hypotheses suggest end. The bend between the Hawaiian and .Emperor Chains that primitive undegassed mantle material ascends beneath reflects a major change in Pacific plate motion at 43.1 ± 1.4 Ma Hawaii. Petrologic models suggest that this primitive material and probably was caused by collision of the Indian subcontinent reacts with the ocean lithosphere to produce the compositional into Eurasia and the resulting reorganization of oceanic spread range of Hawaiian lava. ing centers and initiation of subduction zones in the western Pacific. The volcanoes of the chain were erupted onto the floor of the Pacific Ocean without regard for the age or preexisting INTRODUCTION structure of the ocean crust. Hawaiian volcanoes erupt lava of distinct chemical com The Hawaiian Islands; the seamounts, hanks, and islands of positions during four major stages in their evolution and the Hawaiian Ridge; and the chain of Emperor Seamounts form an growth. -
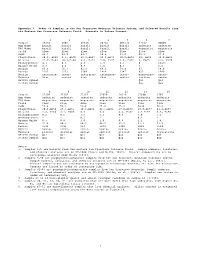
1 Appendix 3. Modes of Samples in the San Francisco Mountain
Appendix 3. Modes of Samples in the San Francisco Mountain Volcanic System, and Selected Basalts from the Eastern San Francisco Volcanic Field. Minerals in Volume Percent. 1 2 3 4 5 6 7 Sample 3823A 2A06 2812A 2031A 2031.B 3732J DC04B Map Name basalt basalt basalt basalt basalt andesite andesite TAS Name basalt basalt basalt basalt basalt mugearite mugearite Field flow flow flow flow flow flow flow SiO2 48.7 48.9 49.7 50.4 51.0 52.2 53.0 Plagioclase 14.6, An69 8.3, An70 25.6,An67 25.5,An71 20.2,An74 29, An64 32.0,An62 Olivine 11.2, Fo83 10.9,Fo84 8.1, Fo73 7.0, Fo77 3.9, Fo76 3, Fo75 6.6, Fo75 Clinopyroxene 4.3 4.3 4.6 1.8 1.3 0 trace Opaque Oxide 4.1 0.2 0.5 1.6 0.3 1 2.4 Matrix 65.8 76.3 61.2 64.1 74.4 67 59.0 Points 1165 1173 1504 1464 1347 827 1549 Matrix intergran- inter- intergran- intergran- inter- microcrys- inter- Texture ular sertal ular ular sertal talline sertal MF1959 Symbol Qa1 Qa1 I-1663 Symbol Qao Qao 8 9 10 11 12 13 14 Sample 3732K 3732Z 3732Y 2705D 3831A 3729Q 3707 Map Name andesite andesite andesite andesite andesite andesite andesite TAS Name mugearite mugearite mugearite mugearite mugearite mugearite mugearite Field flow flow dike flow flow flow flow SiO2 53.4 53.7 53.8 55.2 55.3 56.0 56.6 Plagioclase 24.8,An64 15.3,An61 30.2,An61 42.1,An52 27.0,An62 29.8,An57 41.2,An57 Olivine 2.8, Fo69 3.1, Fo65 4.1 5.6 4.3, Fo62 3.7 Fo57 2.5, Fo58 Clinopyroxene 0.2 0.5 1.5 2.1 0.8 3.0 1.1 Orthopyroxene - - 0.5 - - 0.2 - Opaque Oxide 0.3 0.8 3.0 1.0 0.6 2.1 1.4 Matrix 71.8 80.3 60.7 49.2 67.2 61.1 53.8 Points 1196 1785 854 1844 1428 1029 1540 Matrix hyalo- inter- inter- hyalo- hyalo- hyalo- micro- Texture pilitic granular sertal ophitic pilitic ophitic crystalline MF1959 Symbol Qa1 Qa1 Qai Qa1 Qa2 Qa2 Qa2 I-1663 Symbol Qao Qao Qai Qao Qay Qay Qay Notes: 1. -

Geologic Map of the State of Hawai 'I
Geologic Map of the State of Hawai‘i By David R. Sherrod, John M. Sinton, Sarah E. Watkins, and Kelly M. Brunt Open-File Report 2007–1089 U.S. Department of the Interior U.S. Geological Survey U.S. Department of the Interior DIRK KEMPTHORNE, Secretary U.S. Geological Survey Mark D. Myers, Director U.S. Geological Survey, Reston, Virginia 2007 For product and ordering information: World Wide Web: http://www.usgs.gov/pubprod Telephone: 1-888-ASK-USGS For more information on the USGS—the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment: World Wide Web: http://www.usgs.gov Telephone: 1-888-ASK-USGS Suggested citation: Sherrod, D.R., Sinton, J.M., Watkins, S.E., and Brunt, K.M., 2007, Geologic Map of the State of Hawai`i: U.S. Geological Survey Open-File Report 2007-1089, 83 p., 8 plates, scales 1:100,000 and 1:250,000, with GIS database Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Although this report is in the public domain, permission must be secured from the individual copyright owners to reproduce any copyrighted material contained within this report. ii Geologic Map of the State of Hawai‘i By David R. Sherrod, John M. Sinton, Sarah E. Watkins, and Kelly M. Brunt About this map Sources of mapping, methods of This geologic map and its digital databases present compilation, origin of stratigraphic the geology of the eight major islands of the State of names, and divisions of the geologic Hawai‘i. -
Magmas Parental to the Chassigny Meteorite: New Considerations
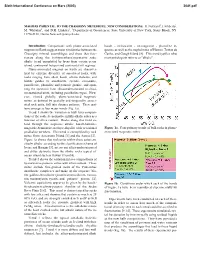
Sixth International Conference on Mars (2003) 3041.pdf MAGMAS PARENTAL TO THE CHASSIGNY METEORITE: NEW CONSIDERATIONS. H. Nekvasil1, J. Filiberto1, M. Whitaker1, and D.H. Lindsley1, 1Department of Geosciences, State University of New York, Stony Brook, NY 11794-2100, [email protected]. Introduction: Comparisons with plume-associated basalt - ne-hawaiite - ne-mugearite - phonolite se- magmas on Earth suggest major similarities between the quence as well as the nephelinites of Hawaii, Tristan da Chassigny mineral assemblages and those that frac- Cunha, and Gough Island [8]. This trend typifies what tionate along the hy(hypersthene)-normative sodic most petrologists refer to as "alkalic". alkalic trend exemplified by lavas from certain ocean island, continental hotspot and continental rift regimes. Plume-associated magmas on Earth are character- ized by extreme diversity of associated rocks, with rocks ranging from alkali basalt, olivine tholeiite, and biotite gabbro to anorthosite, syenite, comendite, pantellerite, phonolite and potassic granite, and span- ning the spectrum from silica-undersaturated to silica- oversaturated units, including peralkaline types. How- ever, viewed globally, plume-associated magmatic suites, as defined by spatially and temporally associ- ated rock units, fall into distinct patterns. These pat- terns emerge as four major trends (Fig. 1a). Trend 1 shows the variation in bulk lava composi- tions of the sodic hy-normative mildly alkalic suites as a function of silica content. Rocks along this trend ex- tend through the sequence alkalic basalt-hawaiite- mugearite-benmoreite-trachyte-rhyolite with occasional Figure 1a. Four primary trends of bulk rocks in plume- peralkaline members. This trend is exemplified by rock associated magmatic suites. -

Origin and Emplacement of a Composite Benmoreite-Trachyte
. I Origin and emplacement of a composite benmoreite - trachyte intrusion: Castle Rock, Lytt~lton Volcano A thesis submitted in partial fulfilment of the requirements for the degree of Master of Science in Geology at the University of Canterbury by Rose Fitzgerald University of Canterbury 1991 II " .. a remarkable dyke forms the crest of the hill, the almost vertical sides of which are in marked contrast with the rounded slopes immediately below it." (Dobson, 1880). I I I Abstract Castle Rock and the Eastern Sheet are elongate intrusions associated with the Lyttelton Volcano, Banks Peninsula. These intrusions were emplaced about 9.7 Ma, late in the evolutionary history of the volcano, and are connected to feeder dikes of the Lyttelton radial swarm. Trachytes, Groups A and B, and benmoreites, low silica and high silica, are components of Castle Rock, whereas only Group A trachyte is found within the Eastern Sheet. The flow foliation and cooling joints of the trachytes and benmoreites indicate the emplacement of bodies with domal cross - sectional shape in a single episode. Velocity gradients in the magmas during emplacement produced groundmass crenulations subsequent to the development of a strong preferred orientation. These crenulations enabled the growing· domes to continuously change their shapes, thus facilitating magma movement within. During the emplacement of Castle Rock magma flow was generally away from the magma source, however, restricted magma movement at the northern end during the trachyte intrusion produced magma movement back towards its origin. Overriding of the trachyte and benmoreite magmas at the advancing magma front did not occur during the emplacement of Castle Rock. -

Lithologic, Age Group, Magnetopolarity, and Geochemical Maps of the Springerville Volcanic Field, East-Central Arizona
U.S. DEPARTMENT OF THE INTERIOR TO ACCOMPANY MAP I–2431 U.S. GEOLOGICAL SURVEY LITHOLOGIC, AGE GROUP, MAGNETOPOLARITY, AND GEOCHEMICAL MAPS OF THE SPRINGERVILLE VOLCANIC FIELD, EAST-CENTRAL ARIZONA By Christopher D. Condit, Larry S. Crumpler, and Jayne C. Aubele INTRODUCTION nantly of monogenetic cinder cones and their asso- ciated flows. The field within Arizona encompasses SETTING AND GENERAL CHARACTERISTICS about 3,000 km2 and has a volume of about 300 OF THE FIELD km3; it contains approximately 400 cones (Condit and others, 1989b). An estimated 100 km2 of the The Springerville volcanic field is one of the many field extends eastward into New Mexico. late Pliocene to Holocene, mostly basaltic, volcanic The geology of the field’s 2,166 km2 of volca- fields present near the Colorado Plateau margin (fig. nic outcrop in Arizona was mapped at 1:24,000 scale, 1 1) . The field overlies the lithospheric transition zone compiled at 1:50,000 scale, and reduced to a scale between the Colorado Plateau and the Basin and of 1:100,000. The south-central part of the field Range Province (Condit and others, 1989b). Estab- (fig. 5, sheet 1) was not mapped because of access lishing relations in time, space, and composition of problems, and detailed mapping in the central part the rocks of these plateau-margin fields offers the of the field extends as far north as about lat 34°27' possibility to integrate more fully into a regional syn- N. Reconnaissance suggests that an additional 50 thesis the detailed geochemistry of these fields now km2 to the north is also covered by flows, a large being examined (for example, Perry and others, 1987; part of which are diktytaxitic; sampling by Cooper Fitton and others, 1988; Menzies and others, 1991). -

Baossi–Warack Monogenetic Volcanoes, Adamawa Plateau, Cameroon: Petrography, Mineralogy and Geochemistry
Acta Geochim (2019) 38(1):40–67 https://doi.org/10.1007/s11631-018-0272-9 ORIGINAL ARTICLE Baossi–Warack monogenetic volcanoes, Adamawa Plateau, Cameroon: petrography, mineralogy and geochemistry 1,2 2 3 Anicet Feudjio Tiabou • Robert Temdjim • Pierre Wandji • 4 1,5 6 Jacques-Marie Bardintzeff • Vivian Bih Che • Edith Ekatah Bate Tibang • 7 7 Caroline Neh Ngwa • Franc¸ois Xavier Onana Mebara Received: 19 December 2017 / Revised: 2 April 2018 / Accepted: 13 April 2018 / Published online: 23 April 2018 Ó Science Press, Institute of Geochemistry, CAS and Springer-Verlag GmbH Germany, part of Springer Nature 2018 Abstract Three monogenetic cones in the Baossi–Warack include titano-magnetite and titano-hematite, nepheline, area, Ngaounde´re´, Adamawa Plateau forming part of the apatite and amphibole xenocrysts. Sanidine occurs in some Cameroon Volcanic Line (CVL) are documented in this samples and sodi-potassic albite in others. Some olivines and study. Basaltic lavas (\ 1km3) scattered around these vents clinopyroxenes exhibit resorbed margins and thin reaction and restricted volcaniclastic deposits were emplaced by rims while plagioclase displays oscillatory zoning, and Hawaiian and mild strombolian style eruptions. The lavas are sieved textures as a result of magma mixing. Whole-rock porphyritic, mainly composed of olivine (chrysolite) and geochemistry data indicates that the lavas are silica-under- clinopyroxene (diopside and augite) phenocrysts and pla- saturated, composed of basanites and basalts, showing little gioclase (andesine) microphenocrysts. Accessory minerals compositional variations (SiO2: 39.20 wt.%–48.01 wt.%, MgO: 5.29 wt.%–9.70 wt.%). Trace elements patterns of these lavas suggest they are enriched in LILE including Pb, Pierre Wandji: Deceased. -

REVISION 1 Intra-Eruptive Trachyte
1 REVISION 1 2 Intra-eruptive trachyte-phonolite transition: natural evidences and 3 experimental constraints on the role of crystal mushes 4 5 Marco Brenna1*, Alessio Pontesilli1, Silvio Mollo2, Matteo Masotta3, Shane J. Cronin4, Ian 6 E.M. Smith4, Mohammed Rashad Hassan Moufti5, Piergiorgio Scarlato6 7 8 1 Geology Department, University of Otago, Dunedin, New Zealand 9 2 Dipartimento di Scienze della Terra, Sapienza Università di Roma, Rome, Italy 10 3 Dipartimento di Scienze della Terra, Università di Pisa, Pisa, Italy 11 4 School of Environment, University of Auckland, Auckland, New Zealand 12 5 Faculty of Earth Sciences, King Abdulaziz University, Jeddah, Saudi Arabia 13 6 Istituto Nazionale di Geofisica e Vulcanologia, Roma, Italy 14 * corresponding author: [email protected] 15 16 ABSTRACT 17 The generation of silica undersaturated phonolite from silica saturated trachytes is 18 uncommon, as it implies the crossing of the thermal barrier and critical plane of silica 19 undersaturation. Nevertheless, a co-genetic suite displaying compositional transition from 20 benmoreite-trachyte to phonolite has been observed within the Al Shaatha pyroclastic 21 sequence in the Harrat Rahat Volcanic Field (Kingdom of Saudi Arabia). We performed 22 crystallization experiments on benmoreite and trachyte starting compositions in order to 23 simulate the pressure-temperature-volatile conditions that generated the observed liquid line 24 of descent. The experimental conditions were 200-500 MPa, 850-1,150 °C, 0-10 wt.% H2O, 25 0.0-0.5 wt.% CO2 and NNO+2 oxygen buffer. The experimental mineral assemblage consists 26 of clinopyroxene, feldspar and titanomagnetite, as well as glass in variable proportions. -

Hawaii Volcanoes National Park Geologic Resources
National Park Service U.S. Department of the Interior Natural Resource Program Center Hawai‘i Volcanoes National Park Geologic Resources Inventory Report Natural Resource Report NPS/NRPC/GRD/NRR—2009/163 THIS PAGE: Geologists have long been monitoring the volcanoes of Hawai‘i Volcanoes National Park.k. Here lava cascades during the 1969-1971 Mauna Ulu eruption of Kīlauea Vollcano. Note the Mauna Ullu fountain in tthee background. U.S. Geological Survey Photo by J. B. Judd (12/30/1969). ON THE COVER: Continuously erupting since 1983, Kīllaueaauea Vollcanocano continues to shape Hawai‘i Volcanoes National Park. Photo courtesy Lisa Venture/University of Cincinnati. Hawai‘i Volcanoes National Park Geologic Resources Inventory Report Natural Resource Report NPS/NRPC/GRD/NRR—2009/163 Geologic Resources Division Natural Resource Program Center P.O. Box 25287 Denver, Colorado 80225 December 2009 U.S. Department of the Interior National Park Service Natural Resource Program Center Denver, Colorado The National Park Service, Natural Resource Program Center publishes a range of reports that address natural resource topics of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate high-priority, current natural resource management information with managerial application. The series targets a general, diverse audience, and may contain NPS policy considerations or address sensitive issues of management applicability. All manuscripts in the series receive the appropriate level of peer review to ensure that the information is scientifically credible, technically accurate, appropriately written for the intended audience, and designed and published in a professional manner. -

Pu'ukoholā Heiau National Historic Site
National Park Service U.S. Department of the Interior Natural Resource Program Center Pu‘ukoholā Heiau National Historic Site Geologic Resources Inventory Report Natural Resource Report NPS/NRPC/GRD/NRR—2011/386 ON THE COVER Pu‘ukoholā Heiau at sunset with Mauna Kea in background (right) and Pelekane Bay in foreground. National Park Service photograph by Greg Cunningham. THIS PAGE Aerial View of Pu‘ukoholā Heiau and Mailekini Heiau (left) looking north towards Kawaihae Harbor and the North Kohala Coast. The gently-sloping flank of Kohala Volcano looms over the landscape. National Park Service photograph by Pierre Lesage. National Park Service photographs courtesy Greg Cunningham (Pu‘ukoholā Heiau National Historic Site). Pu‘ukoholā Heiau National Historic Site Geologic Resources Inventory Report Natural Resource Report NPS/NRPC/GRD/NRR—2011/386 National Park Service Geologic Resources Division PO Box 25287 Denver, CO 80225 April 2011 U.S. Department of the Interior National Park Service Natural Resource Program Center Fort Collins, Colorado The National Park Service, Natural Resource Program Center publishes a range of reports that address natural resource topics of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate high-priority, current natural resource management information with managerial application. The series targets a general, diverse audience, and may contain NPS policy considerations or address sensitive issues of management applicability. All manuscripts in the series receive the appropriate level of peer review to ensure that the information is scientifically credible, technically accurate, appropriately written for the intended audience, and designed and published in a professional manner.