Fetal Megacystis: a Lot More Than LUTO
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Orphanet Report Series Rare Diseases Collection
Orphanet Report Series Rare Diseases collection January 2013 Disease Registries in Europe www.orpha.net 20102206 Table of contents Methodology 3 List of rare diseases that are covered by the listed registries 4 Summary 13 1- Distribution of registries by country 13 2- Distribution of registries by coverage 14 3- Distribution of registries by affiliation 14 Distribution of registries by country 15 European registries 38 International registries 41 Orphanet Report Series - Disease Registries in Europe - January 2013 2 http://www.orpha.net/orphacom/cahiers/docs/GB/Registries.pdf Methodology Patient registries and databases constitute key instruments to develop clinical research in the field of rare diseases (RD), to improve patient care and healthcare planning. They are the only way to pool data in order to achieve a sufficient sample size for epidemiological and/or clinical research. They are vital to assess the feasibility of clinical trials, to facilitate the planning of appropriate clinical trials and to support the enrolment of patients. Registries of patients treated with orphan drugs are particularly relevant as they allow the gathering of evidence on the effectiveness of the treatment and on its possible side effects, keeping in mind that marketing authorisation is usually granted at a time when evidence is still limited although already somewhat convincing. This report gather the information collected by Orphanet so far, regarding systematic collections of data for a specific disease or a group of diseases. Cancer registries are listed only if they belong to the network RARECARE or focus on a rare form of cancer. The report includes data about EU countries and surrounding countries participating to the Orphanet consortium. -

With a Learning Disability; Guidance for General Practice
Improving identification of people with a learning disability: guidance for general practice NHS England and Improvement Publishing Approval Reference: 001030 Version 1 NHS England and NHS Improvement Contents Introduction .................................................................................... 2 Actions for practices ....................................................................... 4 Appendix 1: List of codes that indicate a learning disability ........... 7 Appendix 2: List of codes that may indicate a learning disability . 14 Appendix 3: List of outdated codes .............................................. 20 Appendix 4: Learning disability identification check-list ............... 22 1 | Contents Introduction 1. The NHS Long Term Plan1 commits to improve uptake of the existing annual health check in primary care for people aged over 14 years with a learning disability, so that at least 75% of those eligible have a learning disability health check each year. 2. There is also a need to increase the number of people receiving the annual seasonal flu vaccination, given the level of avoidable mortality associated with respiratory problems. 3. In 2017/18, only 44.6% of patients with a learning disability received a flu vaccination and only 55.1% of patients with a learning disability received an annual learning disability health check.2 4. In June 2019, NHS England and NHS Improvement announced a series of measures to improve coverage of annual health checks and flu vaccination for people with a learning disability. One of the commitments was to improve the quality of registers for people with a learning disability3. Clinical coding review 5. Most GP practices have developed a register of their patients known to have a learning disability. This has been developed from clinical diagnoses, from information gathered from learning disabilities teams and social services and has formed the basis of registers for people with learning disability developed for the Quality and Outcomes Framework (QOF). -

Craniosynostosis Precision Panel Overview Indications Clinical Utility
Craniosynostosis Precision Panel Overview Craniosynostosis is defined as the premature fusion of one or more cranial sutures, often resulting in abnormal head shape. It is a developmental craniofacial anomaly resulting from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). As well, craniosynostosis can be simple when only one suture fuses prematurely or complex/compound when there is a premature fusion of multiple sutures. Complex craniosynostosis are usually associated with other body deformities. The main morbidity risk is the elevated intracranial pressure and subsequent brain damage. When left untreated, craniosynostosis can cause serious complications such as developmental delay, facial abnormality, sensory, respiratory and neurological dysfunction, eye anomalies and psychosocial disturbances. In approximately 85% of the cases, this disease is isolated and nonsyndromic. Syndromic craniosynostosis usually present with multiorgan complications. The Igenomix Craniosynostosis Precision Panel can be used to make a directed and accurate diagnosis ultimately leading to a better management and prognosis of the disease. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. Indications The Igenomix Craniosynostosis Precision Panel is indicated for those patients with a clinical diagnosis or suspicion with or without the following manifestations: ‐ Microcephaly ‐ Scaphocephaly (elongated head) ‐ Anterior plagiocephaly ‐ Brachycephaly ‐ Torticollis ‐ Frontal bossing Clinical Utility The clinical utility of this panel is: - The genetic and molecular confirmation for an accurate clinical diagnosis of a symptomatic patient. - Early initiation of treatment in the form surgical procedures to relieve fused sutures, midface advancement, limited phase of orthodontic treatment and combined 1 orthodontics/orthognathic surgery treatment. -

Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders
177 Arch Neuropsychitry 2020;57:177−191 RESEARCH ARTICLE https://doi.org/10.29399/npa.24890 Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders Hakan GÜRKAN1 , Emine İkbal ATLI1 , Engin ATLI1 , Leyla BOZATLI2 , Mengühan ARAZ ALTAY2 , Sinem YALÇINTEPE1 , Yasemin ÖZEN1 , Damla EKER1 , Çisem AKURUT1 , Selma DEMİR1 , Işık GÖRKER2 1Faculty of Medicine, Department of Medical Genetics, Edirne, Trakya University, Edirne, Turkey 2Faculty of Medicine, Department of Child and Adolescent Psychiatry, Trakya University, Edirne, Turkey ABSTRACT Introduction: Aneuploids, copy number variations (CNVs), and single in 39 (39/123=31.7%) patients. Twelve CNV variant of unknown nucleotide variants in specific genes are the main genetic causes of significance (VUS) (9.75%) patients and 7 CNV benign (5.69%) patients developmental delay (DD) and intellectual disability disorder (IDD). were reported. In 6 patients, one or more pathogenic CNVs were These genetic changes can be detected using chromosome analysis, determined. Therefore, the diagnostic efficiency of CMA was found to chromosomal microarray (CMA), and next-generation DNA sequencing be 31.7% (39/123). techniques. Therefore; In this study, we aimed to investigate the Conclusion: Today, genetic analysis is still not part of the routine in the importance of CMA in determining the genomic etiology of unexplained evaluation of IDD patients who present to psychiatry clinics. A genetic DD and IDD in 123 patients. diagnosis from CMA can eliminate genetic question marks and thus Method: For 123 patients, chromosome analysis, DNA fragment analysis alter the clinical management of patients. Approximately one-third and microarray were performed. Conventional G-band karyotype of the positive CMA findings are clinically intervenable. -

Statistical Analysis Plan
Cover Page for Statistical Analysis Plan Sponsor name: Novo Nordisk A/S NCT number NCT03061214 Sponsor trial ID: NN9535-4114 Official title of study: SUSTAINTM CHINA - Efficacy and safety of semaglutide once-weekly versus sitagliptin once-daily as add-on to metformin in subjects with type 2 diabetes Document date: 22 August 2019 Semaglutide s.c (Ozempic®) Date: 22 August 2019 Novo Nordisk Trial ID: NN9535-4114 Version: 1.0 CONFIDENTIAL Clinical Trial Report Status: Final Appendix 16.1.9 16.1.9 Documentation of statistical methods List of contents Statistical analysis plan...................................................................................................................... /LQN Statistical documentation................................................................................................................... /LQN Redacted VWDWLVWLFDODQDO\VLVSODQ Includes redaction of personal identifiable information only. Statistical Analysis Plan Date: 28 May 2019 Novo Nordisk Trial ID: NN9535-4114 Version: 1.0 CONFIDENTIAL UTN:U1111-1149-0432 Status: Final EudraCT No.:NA Page: 1 of 30 Statistical Analysis Plan Trial ID: NN9535-4114 Efficacy and safety of semaglutide once-weekly versus sitagliptin once-daily as add-on to metformin in subjects with type 2 diabetes Author Biostatistics Semaglutide s.c. This confidential document is the property of Novo Nordisk. No unpublished information contained herein may be disclosed without prior written approval from Novo Nordisk. Access to this document must be restricted to relevant parties.This -

Omphalocele and Gastroschisis Precision Panel Overview Indications Clinical Utility
Omphalocele and Gastroschisis Precision Panel Overview Omphalocele, also known as exomphalos, is a midline abdominal wall defect at the base of the umbilical cord where herniation of abdominal contents takes place. The herniated organs are covered by the parietal peritoneum. The cause of omphalocele postulated to be a failure of the bowel to return into the abdomen by 10-12 weeks. Omphaloceles are associated with other anomalies in more than 70% of the cases, generally chromosomal, and the severity is dictated by the anomalies that are present. The main difficulty of this condition is the exclusion of associated conditions, not all diagnosed prenatally. Gastroschisis represents a herniation of abdominal contents through a paramedian full-thickness abdominal fusion defect. The abdominal herniation, in contrast with omphalocele, is usually to the right of the umbilical cord. It usually contains small bowel and has no surrounding membrane. Challenges in management of gastroschisis are related to the prevention of late intrauterine death, and the prediction and treatment of complex forms. The Igenomix Omphalocele and Gastroschisis Gene Panel can be used to as a screening tool for underlying genetic alterations associated to these conditions. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. Indications The Igenomix Omphalocele and Gastroschisis Gene Panel is indicated for those patients with a clinical suspicion of omphalocele and/or gastroschisis which manifest as: - Herniation of intestines through abdominal wall - Polyhydramnios in utero - Elevated levels of maternal serum a-fetoprotein (MSAFP) Clinical Utility The clinical utility of this panel is: - The genetic and molecular confirmation for an accurate clinical diagnosis of a symptomatic patient. -

EUROCAT Syndrome Guide
JRC - Central Registry european surveillance of congenital anomalies EUROCAT Syndrome Guide Definition and Coding of Syndromes Version July 2017 Revised in 2016 by Ingeborg Barisic, approved by the Coding & Classification Committee in 2017: Ester Garne, Diana Wellesley, David Tucker, Jorieke Bergman and Ingeborg Barisic Revised 2008 by Ingeborg Barisic, Helen Dolk and Ester Garne and discussed and approved by the Coding & Classification Committee 2008: Elisa Calzolari, Diana Wellesley, David Tucker, Ingeborg Barisic, Ester Garne The list of syndromes contained in the previous EUROCAT “Guide to the Coding of Eponyms and Syndromes” (Josephine Weatherall, 1979) was revised by Ingeborg Barisic, Helen Dolk, Ester Garne, Claude Stoll and Diana Wellesley at a meeting in London in November 2003. Approved by the members EUROCAT Coding & Classification Committee 2004: Ingeborg Barisic, Elisa Calzolari, Ester Garne, Annukka Ritvanen, Claude Stoll, Diana Wellesley 1 TABLE OF CONTENTS Introduction and Definitions 6 Coding Notes and Explanation of Guide 10 List of conditions to be coded in the syndrome field 13 List of conditions which should not be coded as syndromes 14 Syndromes – monogenic or unknown etiology Aarskog syndrome 18 Acrocephalopolysyndactyly (all types) 19 Alagille syndrome 20 Alport syndrome 21 Angelman syndrome 22 Aniridia-Wilms tumor syndrome, WAGR 23 Apert syndrome 24 Bardet-Biedl syndrome 25 Beckwith-Wiedemann syndrome (EMG syndrome) 26 Blepharophimosis-ptosis syndrome 28 Branchiootorenal syndrome (Melnick-Fraser syndrome) 29 CHARGE -
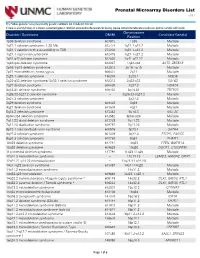
Prenatal Microarray Disorders List V19.1
Prenatal Microarray Disorders List v19.1 This "whole genome" array may identify genetic conditions not included in this list. If there is a family history of a known suspected genetic condition unrelated to the reason for testing, please contact the laboratory to discuss prior to sample submission. Chromosome Disorder / Syndrome OMIM Candidate Gene(s) Position 1p36 deletion syndrome 607872 1p36 Multiple 1q21.1 deletion syndrome, 1.35 Mb 612474 1q21.1-q21.2 Multiple 1q21.1 deletion with susceptibility to TAR 274000 1q21.1-q21.2 Multiple 1q21.1 duplication syndrome 612475 1q21.1-q21.2 Multiple 1q41-q42 deletion syndrome 612530 1q41-q42.12 Multiple 1q43-q44 deletion syndrome 612337 1q43-q44 AKT3, ZBTB18 2p16.1-p15 deletion syndrome 612513 2p16.1-p15 Multiple 2p21 microdeletion, homozygous 606407 2p21 Multiple 2q23.1 deletion syndrome 156200 2q23.1 MBD5 2q32-q33 deletion syndrome/ 2q33.1 deletion syndrome 612313 2q32-q33 SATB2 2q37 deletion syndrome 600430 2q37.3 HDAC4 3q13.31 deletion syndrome 615433 3q13.31 ZBTB20 3q26.33-3q27.2 deletion syndrome -- 3q26.33-3q27.2 Multiple 3q27.3 deletion syndrome -- 3q27.3 Multiple 3q29 deletion syndrome 609425 3q29 Multiple 4q21 deletion syndrome 613509 4q21 Multiple 5q14.3 deletion syndrome 613443 5q14.3 MEF2C 6pter-p24 deletion syndrome 612582 6pter-p24 Multiple 7q11.23 distal deletion syndrome 613729 7q11.23 Multiple 7q11.23 duplication syndrome 609757 7q11.23 Multiple 8p23.1 deletion/duplication syndrome 600576 8p23.1 GATA4 9q22.3 deletion syndrome 601309 9q22.3 PTCH1, FANCC 9q34.3 deletion syndrome -

Syndromes and Constitutional Chromosomal Abnormalities
Downloaded from jmg.bmj.com on May 16, 2013 - Published by group.bmj.com 705 REVIEW Syndromes and constitutional chromosomal abnormalities associated This article is available free on JMG online via the JMG Unlocked open access trial, with Wilms tumour funded by the Joint Information Systems Committee. For further information, see R H Scott, C A Stiller, L Walker, N Rahman http://jmg.bmjjournals.com/cgi/content/ full/42/2/97 ............................................................................................................................... J Med Genet 2006;43:705–715. doi: 10.1136/jmg.2006.041723 Wilms tumour has been reported in association with over Multiple nephrogenic rests throughout the kid- ney are sometimes present and can be referred to 50 different clinical conditions and several abnormal as nephroblastomatosis. constitutional karyotypes. Conclusive evidence of an The dramatic advances in the treatment of increased risk of Wilms tumour exists for only a minority of Wilms tumour are such that long term survival is now the norm: exceeding 90% for disease these conditions, including WT1 associated syndromes, localised to the abdomen (stages 1–3), and over familial Wilms tumour, and certain overgrowth conditions 70% in metastatic disease (stage 4).7 The treat- such as Beckwith-Wiedemann syndrome. In many reported ment of Wilms tumour is determined both by stage and histological classification of the conditions the rare co-occurrence of Wilms tumour is tumour, with treatment protocols varying probably due to chance. However, for several conditions between countries. Surgical resection is comple- the available evidence cannot either confirm or exclude an mented in the majority of individuals by chemo- therapy. Currently in the United Kingdom increased risk, usually because of the rarity of the around 30% of individuals with Wilms tumour syndrome. -

Genetic Mapping and Mutation Analysis of Genes Causing Human Hereditary Skeletal Disorders
Genetic Mapping and Mutation Analysis of Genes Causing Human Hereditary Skeletal Disorders by Irfan Ullah Department of Biochemistry Faculty of Biological Sciences Quaid-i-Azam University Islamabad, Pakistan Session: 2012-2017 Genetic Mapping and Mutation Analysis of Genes Causing Human Hereditary Skeletal Disorders A thesis submitted in the partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biochemistry by Irfan Ullah Department of Biochemistry Faculty of Biological Sciences Quaid-i-Azam University Islamabad, Pakistan Session: 2012-2017 Dedicated to Those marvelous personalities Whose Love is Ceaseless Whose Affections are Limitless Whose Compassions are Matchless and Whose prayers are Selfless Incontrovertibly They are my kind father and sweet Mother May they live long! Contents CONTENTS Page No ACKNOWLEDGMENTS I LIST OF FIGURES III LIST OF TABLES IX LIST OF ABBREVIATIONS X ABSTRACT XVII Chapter 1 INTRODUCTION 1 Human Skeletal Architecture 2 The Skeletal Paraphernalia 4 Bone 4 Histology of Bone 4 Bone Anatomy 4 Types of Bones 5 Cartilage 6 Types of Cartilages 6 Skeletal Development 6 Genetic Regulation of Skeletal Development 8 Condensation of Mesenchymal Cells and Skeletal Patterning 8 Chondrocyte Differentiation and Development of Cartilage Growth Plate 10 Osteoblast differentiation and Bone Development 11 Osteoclast Differentiation and Bone Homeostasis 13 Hereditary Skeletal Disorders 13 Acromesomelic Dysplasias 14 Mucopolysaccharidosis 15 Ciliopathic Disorders of the Skeleton 18 Hereditary Limb Malformations -

Ocular Coloboma: a Reassessment in the Age of Molecular Neuroscience
881 REVIEW Ocular coloboma: a reassessment in the age of molecular neuroscience C Y Gregory-Evans, M J Williams, S Halford, K Gregory-Evans ............................................................................................................................... J Med Genet 2004;41:881–891. doi: 10.1136/jmg.2004.025494 Congenital colobomata of the eye are important causes of NORMAL EYE DEVELOPMENT The processes that occur during formation of the childhood visual impairment and blindness. Ocular vertebrate eye are well documented and include coloboma can be seen in isolation and in an impressive (i) multiple inductive and morphogenetic events, number of multisystem syndromes, where the eye (ii) proliferation and differentiation of cells into mature tissue, and (iii) establishment of neural phenotype is often seen in association with severe networks connecting the retina to the higher neurological or craniofacial anomalies or other systemic neural centres such as the superior colliculus, the developmental defects. Several studies have shown that, in geniculate nucleus, and the occipital lobes.8–10 At around day 30 of gestation, the ventral surface addition to inheritance, environmental influences may be of the optic vesicle and stalk invaginates leading causative factors. Through work to identify genes to the formation of a double-layered optic cup. underlying inherited coloboma, significant inroads are This invagination gives rise to the optic fissure, allowing blood vessels from the vascular meso- being made into understanding the molecular events derm to enter the developing eye. Fusion of the controlling closure of the optic fissure. In general, severity edges of this fissure starts centrally at about of disease can be linked to the temporal expression of the 5 weeks and proceeds anteriorly towards the rim of the optic cup and posteriorly along the optic gene, but this is modified by factors such as tissue stalk, with completion by 7 weeks.11 Failure of specificity of gene expression and genetic redundancy. -

Genetics and Metabolics Abbreviations and Diagnosis
Genetics and Metabolics Abbreviations and Diagnosis Abbreviation Diagnosis Provider/Specialty 22-Q Deletion Genetics 19p Partial Trisomy Syndrome Genetics 1 P3 Deletion Syndrome Genetics 16q23 Duplication Genetics Aarskog-Scott Syndrome Genetics Abnormal single gene testing-confirmed Genetics/High priority for scheduling Achondroplasia Genetics Acute Reactive Lymphadenopathy Genetic Adrenoleukodystrophy Metabolics Alagille Syndrome Genetics Albinism Genetics Albright-McCune Syndrome Genetics Alkaptonuria Metabolics Ambiguous Genitalia Genetics Amblyopia Genetics Amyoplasia Congenital Genetics Angelman’s Syndrome Genetics Anterior Segment Dysgenesis of Eyes Genetics Apert Syndrome Genetics Apraxia of Speech (verbal apraxia/language Genetics apraxia) Arterio Venous Malformation Genetics Arthrogryposis Genetics Ataxia Genetics Atrophy Genetics Autism including family hx Genetics Axenfeld Syndrome Genetics Azoospermia R/O genetic cause Genetics Information gathered from https://wiki.utah.edu/confluence/pages/viewpage.action?pageId=146113859. Reviewed, edited and approved by Melissa Smith, Metabolic Clinic Coordinator, University of Utah Pediatric Genetics. Updated 05/2016. B12 Vitamin Deficiency Metabolics Barth Syndrome Metabolics BWS Beckwith-Wiedemann Syndrome Genetics Betakethiolase Deficiency Metabolics Bilateral Club Feet Genetics Bilateral Subluxated Lenses Genetics Biotin Deficiency Metabolics Bone Malformation Genetics Brachydactyly Genetics Brain-Thyroid-Lung Syndrome Genetics Brachial Oto Renal Syndrome Genetics CFTR Mutations