Prediction of RNA-Binding Proteins from Primary Sequence by a Support Vector Machine Approach
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

METACYC ID Description A0AR23 GO:0004842 (Ubiquitin-Protein Ligase
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Heat Stress Responsive Zostera marina Genes, Southern Population (α=0. -

WO 2017/014762 Al 26 January 2017 (26.01.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/014762 Al 26 January 2017 (26.01.2017) P O P C T (51) International Patent Classification: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, C12Q 1/68 (2006.01) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (21) International Application Number: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PCT/US201 5/0414 15 PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (22) International Filing Date: SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 2 1 July 20 15 (21 .07.2015) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, (71) Applicant: OMNIOME, INC. [US/US]; 4225 Executive TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, Square, Suite 440, La Jolla, California 92037 (US). TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (72) Inventors: VIJAYAN, Kandaswamy; 4465 Vision Drive, LV, MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, Unit 6, San Diego, California 92121 (US). -

Mapping Active Site Residues in Glutamate-5-Kinase. the Substrate Glutamate and the Feed-Back Inhibitor Proline Bind at Overlapping Sites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 580 (2006) 6247–6253 Mapping active site residues in glutamate-5-kinase. The substrate glutamate and the feed-back inhibitor proline bind at overlapping sites Isabel Pe´rez-Arellanoa, Vicente Rubiob, Javier Cerveraa,* a Centro de Investigacio´n Prı´ncipe Felipe, Avda. Autopista del Saler, 16, Valencia 46013, Spain b Instituto de Biomedicina de Valencia (IBV-CSIC), Jaime Roig 11, Valencia 46010, Spain Received 22 August 2006; revised 10 October 2006; accepted 12 October 2006 Available online 20 October 2006 Edited by Stuart Ferguson (a domain named after pseudo uridine synthases and archaeo- Abstract Glutamate-5-kinase (G5K) catalyzes the controlling first step of proline biosynthesis. Substrate binding, catalysis sine-specific transglycosylases) domain [10]. AAK domains and feed-back inhibition by proline are functions of the N-termi- bind ATP and a phosphorylatable acidic substrate, and make nal 260-residue domain of G5K. We study here the impact on a phosphoric anhydride [11]. The function of the PUA domain these functions of 14 site-directed mutations affecting 9 residues (a domain characteristically found in some RNA-modifying of Escherichia coli G5K, chosen on the basis of the structure of enzymes) [12] is not known. By deleting the PUA domain of the bisubstrate complex of the homologous enzyme acetylgluta- E. coli G5K we showed [10] that the isolated AAK domain, mate kinase (NAGK). The results support the predicted roles as the complete enzyme, forms tetramers, catalyzes the reac- of K10, K217 and T169 in catalysis and ATP binding and of tion and is feed-back inhibited by proline. -

Supplementary Materials
Supplementary Materials Figure S1. Differentially abundant spots between the mid-log phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 3. Figure S2. Differentially abundant spots between the stationary phase cells grown on xylan or xylose. Red and blue circles denote spots with increased and decreased abundance respectively in the xylan growth condition. The identities of the circled spots are summarized in Table 4. S2 Table S1. Summary of the non-polysaccharide degrading proteins identified in the B. proteoclasticus cytosol by 2DE/MALDI-TOF. Protein Locus Location Score pI kDa Pep. Cov. Amino Acid Biosynthesis Acetylornithine aminotransferase, ArgD Bpr_I1809 C 1.7 × 10−4 5.1 43.9 11 34% Aspartate/tyrosine/aromatic aminotransferase Bpr_I2631 C 3.0 × 10−14 4.7 43.8 15 46% Aspartate-semialdehyde dehydrogenase, Asd Bpr_I1664 C 7.6 × 10−18 5.5 40.1 17 50% Branched-chain amino acid aminotransferase, IlvE Bpr_I1650 C 2.4 × 10−12 5.2 39.2 13 32% Cysteine synthase, CysK Bpr_I1089 C 1.9 × 10−13 5.0 32.3 18 72% Diaminopimelate dehydrogenase Bpr_I0298 C 9.6 × 10−16 5.6 35.8 16 49% Dihydrodipicolinate reductase, DapB Bpr_I2453 C 2.7 × 10−6 4.9 27.0 9 46% Glu/Leu/Phe/Val dehydrogenase Bpr_I2129 C 1.2 × 10−30 5.4 48.6 31 64% Imidazole glycerol phosphate synthase Bpr_I1240 C 8.0 × 10−3 4.7 22.5 8 44% glutamine amidotransferase subunit Ketol-acid reductoisomerase, IlvC Bpr_I1657 C 3.8 × 10−16 -

DNA Methylation Seeing the Forest for the Trees: a Wide Perspective on RNA-Directed
Downloaded from genesdev.cshlp.org on August 16, 2012 - Published by Cold Spring Harbor Laboratory Press Seeing the forest for the trees: a wide perspective on RNA-directed DNA methylation Huiming Zhang and Jian-Kang Zhu Genes Dev. 2012 26: 1769-1773 Access the most recent version at doi:10.1101/gad.200410.112 References This article cites 31 articles, 9 of which can be accessed free at: http://genesdev.cshlp.org/content/26/16/1769.full.html#ref-list-1 Related Content Spatial and functional relationships among Pol V-associated loci, Pol IV-dependent siRNAs, and cytosine methylation in the Arabidopsis epigenome Andrzej T. Wierzbicki, Ross Cocklin, Anoop Mayampurath, et al. Genes Dev. August 15, 2012 26: 1825-1836 Email alerting Receive free email alerts when new articles cite this article - sign up in the box at the service top right corner of the article or click here Topic Articles on similar topics can be found in the following collections Collections Chromatin and Gene Expression (146 articles) Plant Biology (29 articles) To subscribe to Genes & Development go to: http://genesdev.cshlp.org/subscriptions Copyright © 2012 by Cold Spring Harbor Laboratory Press Downloaded from genesdev.cshlp.org on August 16, 2012 - Published by Cold Spring Harbor Laboratory Press PERSPECTIVE Seeing the forest for the trees: a wide perspective on RNA-directed DNA methylation Huiming Zhang1 and Jian-Kang Zhu1,2,3 1Department of Horticulture and Landscape Architecture, Purdue University, West Lafayette, Indiana 47907, USA; 2Shanghai Center for Plant Stress Biology, Shanghai Institutes of Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China In this issue of Genes & Development, Wierzbicki and remaining subunits of Pol IV and/or Pol V are different but colleagues (pp. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -
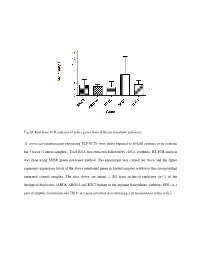
Fig.S1 Real-Time PCR Analysis of Select Genes from Different Metabolic Pathways S. Cerevisiae Transformants Expressing TEF-YCT1
Fig.S1 Real-time PCR analysis of select genes from different metabolic pathways S. cerevisiae transformants expressing TEF-YCT1 were either exposed to 500µM cysteine or no cysteine for 5 hours (Control samples). Total RNA was extracted followed by cDNA synthesis. RT-PCR analysis was done using SYBR green dye-based method. The experiment was carried out twice and the figure represents expression levels of the above mentioned genes in treated samples relative to the corresponding untreated control samples. The data above are means ± SD from technical replicates (n=3) of the biological duplicates. [ARG8, ARG5,6 and RTC2 belong to the arginine biosynthetic pathway, SSU1 is a part of sulphur metabolism and TIS11 is a gene involved in maintaining iron homeostasis in the cells.] Table S1: List of strains used in this study Strain Genotype Source ABC 1738 (yct1) MATα his31 leu20 lys20 ura30 YLL055W:: kanMX4 Euroscarf (Y01543) ABC3612(arg3) MATa his31 leu20 ura30 YJL088w::kanMX4 Euroscarf (Y11335) ABC4812(arg5,6) MATa his31 leu20 ura30 YER069w::kanMX4 Euroscarf (Y10209) ABC4811(arg8) MATa his31 leu20 ura30 YOL140w::kanMX4 Euroscarf (Y17711) ABC4814(spe1) MATa his31 leu20 ura30 YKL184w::kanMX4 Euroscarf (Y15034) ABC4815(spe2) MATa his31 leu20 ura30 YOL052c::kanMX4 Euroscarf (Y11743) ABC4816(spe3) MATa his31 leu20 ura30 YPR069c::kanMX4 Euroscarf (Y15488) ABC4817(spe4) MATa his31 leu20 ura30 YLR146c::kanMX4 Euroscarf (Y16945) ABC3604(ssu1) MATa his31 leu20 ura30 YPL092w::kanMX4 Euroscarf (Y12160) ABC1486(str2) MATa his31 leu20 ura30 YJR130c::kanMX4 -

The Pennsylvania State University
The Pennsylvania State University The Graduate School Graduate Program in Plant Biology IDENTIFICATION OF SMALL RNA PRODUCING GENES IN THE MOSS PHYSCOMITRELLA PATENS A Dissertation in Plant Biology by Ceyda Coruh 2014 Ceyda Coruh Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy August 2014 The dissertation of Ceyda Coruh was reviewed and approved* by the following: Michael J. Axtell Associate Professor of Biology Dissertation Advisor Chair of Committee Claude dePamphilis Professor of Biology Sarah M. Assmann Waller Professor of Biology Anton Nekrutenko Associate Professor of Biochemistry and Molecular Biology Teh-hui Kao Distinguished Professor of Biochemistry and Molecular Biology Chair, Intercollege Graduate Degree Program in Plant Biology *Signatures are on file in the Graduate School iii ABSTRACT In plants, a significant fraction of the genome is responsible for making regulatory small RNAs. These ubiquitous, endogenous small RNAs are currently categorized into two groups: microRNAs (miRNAs) and small interfering RNAs (siRNAs). They are produced by Dicer-Like (DCL) proteins and utilized by Argonaute (AGO) proteins to guide repressive regulation of target mRNAs and/or chromatin selected on the basis of small RNA-target complementarity at the transcriptional or post-transcriptional levels. 21 nt miRNAs and 24 nt heterochromatic siRNAs are the two major types of small RNAs found in angiosperms (flowering plants). The small RNA populations in angiosperms are dominated by 24 nt heterochromatic siRNAs which derive from intergenic, repetitive regions and mediate DNA methylation and repressive histone modifications to targeted loci in angiosperms. However, the existence and extent of heterochromatic siRNAs in other land plant lineages has been less clear. -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

Materials and Methods
Effects of Hybridization on Heterochromatic Small Interfering RNA and Gene Expression in Zea mays by Travis Korry Coleman A Thesis presented to The University of Guelph In partial fulfilment of requirements for the degree of Doctor of Philosophy in Plant Agriculture Guelph, Ontario, Canada © Travis Coleman, January, 2017 ABSTRACT EFFECTS OF HYBRIDIZATION ON HETEROCHROMATIC SMALL INTERFERING RNA AND GENE EXPRESSION IN ZEA MAYS Travis Korry Coleman Advisor: University of Guelph, 2016 Associate Professor Lewis N. Lukens Despite decades of research, the molecular nature of heterosis is not completely understood. Heterosis for quantitative traits is controlled by the cumulative effects of multiple genes and regulatory elements, each of which may exhibit differing modes of action. While dominance and over-dominance theories can account for some of the genetic control of heterosis, neither provides a complete account. There is a growing body of evidence that implicates small RNAs as non-coding elements which may play a role in the manifestation of heterosis upon hybridization. In particular, 24-nt heterochromatic small interfering RNAs (hetsiRNAs) may play a role in mediating trans-genomic interactions via DNA methylation when two genomes come into contact in an F1 nucleus. Recent research in Arabidopsis thaliana has demonstrated that hetsiRNAs show non-additive expression upon hybridization, with the majority of hetsiRNAs being downregulated. This research seeks to examine such trends in non-additive hetsiRNA expression in commercial maize germplasm. Small RNA were isolated and deep sequenced from leaf tissue samples of two inbred lines and their F1 hybrid. In order to examine the effects of reducing hetsiRNAs in hybridization, each genotype was also sampled in each of two mediator of paramutation 1 (mop1) allelic states. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Saccharomyces Cerevisiae Aspartate Kinase Mechanism and Inhibition
In compliance with the Canadian Privacy Legislation some supporting forms may have been removed from this dissertation. While these forms may be included in the document page count, their removal does not represent any loss of content from the dissertation. Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry FUNGAL ASPARTATE KINASE MECHANISM AND INHIBITION By DAVID C. BAREICH, B.Sc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy McMaster University © Copyright by David C. Bareich, June 2003 1 Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry FUNGAL ASPARTATE KINASE MECHANISM AND INHIBITION Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry DOCTOR OF PHILOSOPHY (2003) McMaster University (Biochemistry) Hamilton, Ontario TITLE: Saccharomyces cerevisiae aspartate kinase mechanism and inhibition AUTHOR: David Christopher Bareich B.Sc. (University of Waterloo) SUPERVISOR: Professor Gerard D. Wright NUMBER OF PAGES: xix, 181 11 Ph.D. Thesis - D. Bareich McMaster University - Department of Biochemistry ABSTRACT Aspartate kinase (AK) from Saccharomyces cerevisiae (AKsc) catalyzes the first step in the aspartate pathway responsible for biosynthesis of L-threonine, L-isoleucine, and L-methionine in fungi. Little was known about amino acids important for AKsc substrate binding and catalysis. Hypotheses about important amino acids were tested using site directed mutagenesis to substitute these amino acids with others having different properties. Steady state kinetic parameters and pH titrations of the variant enzymes showed AKsc-K18 and H292 to be important for binding and catalysis. Little was known about how the S. cerevisiae aspartate pathway kinases, AKsc and homoserine kinase (HSKsc), catalyze the transfer of the y-phosphate from adenosine triphosphate (ATP) to L-aspartate or L-homoserine, respectively.