Detection of Viable Yersinia Pestis by Fluorescence in Situ Hybridization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

E. Coli (Expec) Among E
Elucidating the Unknown Ecology of Bacterial Pathogens from Genomic Data Tristan Kishan Seecharran A thesis submitted in partial fulfilment of the requirements of Nottingham Trent University for the degree of Doctor of Philosophy June 2018 Copyright Statement I hereby declare that the work presented in this thesis is the result of original research carried out by the author, unless otherwise stated. No material contained herein has been submitted for any other degree, or at any other institution. This work is an intellectual property of the author. You may copy up to 5% of this work for private study, or personal, non-commercial research. Any re-use of the information contained within this document should be fully referenced, quoting the author, title, university, degree level and pagination. Queries or requests for any other use, or if a more substantial copy is required, should be directed in the owner(s) of the Intellectual Property Rights. Tristan Kishan Seecharran i Acknowledgements I would like to express my sincere gratitude and thanks to my external advisor Alan McNally and director of studies Ben Dickins for their continued support, guidance and encouragement, and without whom, the completion of this thesis would not have been possible. Many thanks also go to the members of the Pathogen Research Group at Nottingham Trent University. I would like to thank Gina Manning and Jody Winter in particular for their invaluable advice and contributions during lab meetings. I would also like to thank our collaborators, Mikael Skurnik and colleagues from the University of Helsinki and Jukka Corander from the University of Oslo, for their much-appreciated support and assistance in this project and the published work on Yersinia pseudotuberculosis. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

Antarctic Rahnella Inusitata: a Producer of Cold-Stable Β-Galactosidase Enzymes
International Journal of Molecular Sciences Article Antarctic Rahnella inusitata: A Producer of Cold-Stable β-Galactosidase Enzymes Kattia Núñez-Montero 1,2,†, Rodrigo Salazar 1,3,†, Andrés Santos 1,3,†, Olman Gómez-Espinoza 2,4 , Scandar Farah 1 , Claudia Troncoso 1,3 , Catalina Hoffmann 1, Damaris Melivilu 1, Felipe Scott 5 and Leticia Barrientos Díaz 1,* 1 Laboratory of Molecular Applied Biology, Center of Excellence in Translational Medicine, Universidad de La Frontera, Avenida Alemania 0458, Temuco 4810296, Chile; [email protected] (K.N.-M.); [email protected] (R.S.); [email protected] (A.S.); [email protected] (S.F.); [email protected] (C.T.); [email protected] (C.H.); [email protected] (D.M.) 2 Biotechnology Investigation Center, Department of Biology, Instituto Tecnológico de Costa Rica, Cartago 159-7050, Costa Rica; [email protected] 3 Scientific and Technological Bioresource Nucleus (BIOREN), Universidad de La Frontera, Temuco 4811230, Chile 4 Laboratory of Plant Physiology and Molecular Biology, Institute of Agroindustry, Department of Agronomic Sciences and Natural Resources, Faculty of Agricultural and Forestry Sciences, Universidad de La Frontera, Temuco 4811230, Chile 5 Green Technology Research Group, Facultad de Ingenieria y Ciencias Aplicadas, Universidad de los Andes, Santiago 7620001, Chile; [email protected] * Correspondence: [email protected]; Tel.: +56-45-259-2802 Citation: Núñez-Montero, K.; † These authors contributed equally to this work. Salazar, R.; Santos, A.; Gómez- Espinoza, O.; Farah, S.; Troncoso, C.; Abstract: There has been a recent increase in the exploration of cold-active β-galactosidases, as it Hoffmann, C.; Melivilu, D.; Scott, F.; offers new alternatives for the dairy industry, mainly in response to the current needs of lactose- Barrientos Díaz, L. -

List of the Pathogens Intended to Be Controlled Under Section 18 B.E
(Unofficial Translation) NOTIFICATION OF THE MINISTRY OF PUBLIC HEALTH RE: LIST OF THE PATHOGENS INTENDED TO BE CONTROLLED UNDER SECTION 18 B.E. 2561 (2018) By virtue of the provision pursuant to Section 5 paragraph one, Section 6 (1) and Section 18 of Pathogens and Animal Toxins Act, B.E. 2558 (2015), the Minister of Public Health, with the advice of the Pathogens and Animal Toxins Committee, has therefore issued this notification as follows: Clause 1 This notification is called “Notification of the Ministry of Public Health Re: list of the pathogens intended to be controlled under Section 18, B.E. 2561 (2018).” Clause 2 This Notification shall come into force as from the following date of its publication in the Government Gazette. Clause 3 The Notification of Ministry of Public Health Re: list of the pathogens intended to be controlled under Section 18, B.E. 2560 (2017) shall be cancelled. Clause 4 Define the pathogens codes and such codes shall have the following sequences: (1) English alphabets that used for indicating the type of pathogens are as follows: B stands for Bacteria F stands for fungus V stands for Virus P stands for Parasites T stands for Biological substances that are not Prion R stands for Prion (2) Pathogen risk group (3) Number indicating the sequence of each type of pathogens Clause 5 Pathogens intended to be controlled under Section 18, shall proceed as follows: (1) In the case of being the pathogens that are utilized and subjected to other law, such law shall be complied. (2) Apart from (1), the law on pathogens and animal toxin shall be complied. -

Epidemiology and Comparative Analysis of Yersinia in Ireland Author(S) Ringwood, Tamara Publication Date 2013 Original Citation Ringwood, T
UCC Library and UCC researchers have made this item openly available. Please let us know how this has helped you. Thanks! Title Epidemiology and comparative analysis of Yersinia in Ireland Author(s) Ringwood, Tamara Publication date 2013 Original citation Ringwood, T. 2013. Epidemiology and comparative analysis of Yersinia in Ireland. PhD Thesis, University College Cork. Type of publication Doctoral thesis Rights © 2013, Tamara Ringwood http://creativecommons.org/licenses/by-nc-nd/3.0/ Item downloaded http://hdl.handle.net/10468/1294 from Downloaded on 2021-10-07T12:07:10Z Epidemiology and Comparative Analysis of Yersinia in Ireland by Tamara Ringwood A thesis presented for the Degree of Doctor of Philosophy National University of Ireland, Cork University College Cork Coláiste na hOllscoile Corcaigh Department of Microbiology Head of Department: Prof. Gerald F. Fitzgerald Supervisor: Prof. Michael B. Prentice April 2013 Contents List of Tables .............................................................................................................................................. iv List of figures ............................................................................................................................................. vi Declaration ............................................................................................................................................... viii Acknowledgements ................................................................................................................................ -

Tierärztliche Hochschule Hannover Untersuchungen Zum Auftreten
Tierärztliche Hochschule Hannover Untersuchungen zum Auftreten verschiedener bakterieller Zoonoseerreger und zu den Risikofaktoren in norddeutschen Schweinemastbeständen INAUGURAL – DISSERTATION zur Erlangung des Grades einer Doktorin oder eines Doktors der Veterinärmedizin - Doctor medicinae veterinariae - ( Dr. med. vet. ) vorgelegt von Susanne Döhne Marburg/Lahn Hannover 2010 Wissenschaftliche Betreuung: Prof. Dr. L. Kreienbrock, Institut für Biometrie, Epidemiologie und Informationsverarbeitung Prof. Dr. K.-H. Waldmann, Klinik für kleine Klauentiere und forensische Medizin und Ambulatorische Klinik 1. Gutachter: Prof. Dr. L. Kreienbrock, Prof. Dr. K.-H. Waldmann 2. Gutachter: Prof. Dr. T. Blaha Tag der mündlichen Prüfung: 03.11.2010 Diese Dissertation wurde gefördert durch das Bundesministerium für Bildung und Forschung im Rahmen des Projektes FBI-Zoo Veröffentlichungen Erste Ergebnisse dieser Arbeit wurden bereits veröffentlicht: DÖHNE, S., A. VON ALTROCK, R. MERLE, K.-H. WALDMANN u. L. KREIENBROCK (2009): Incidence and antimicrobial susceptibility of Salmonella in fattening pig herds in Northern Germany. In: 14 th ISAH Congress Vechta, 19.-23. Juli 2009, Proceedings Vol.2 DÖHNE, S., A. VON ALTROCK, R. MERLE, K.-H. WALDMANN u. L. KREIENBROCK (2009): Resistenzen von Salmonella spp. in norddeutschen Schweinemastbetrieben. In: DVG-Fachgruppentagung Epidemiologie und Dokumentation: „Krankheitsdynamik in Populationen – Bedeutung von Surveillance und Impfprogrammen“, Gießen, 2.-4. September 2009 Inhaltsverzeichnis 1 Einleitung 13 2 Literaturübersicht -

Clinical Isolates of Yersinia Enterocolitica in Finland 117 2013 117
Leila M. Sihvonen Leila Leila M. Sihvonen Clinical isolates of Yersinia enterocolitica in Finland Leila M. Sihvonen Identification and Epidemiology Clinical isolates of Yersinia enterocolitica in Finland Identification and Epidemiology Clinical isolates of Clinical isolates Yersinia enterocolitica is a foodborne bacterium that causes gastroenteritis and post-infectious complications, such as reactive arthritis, in humans. Y. enterocolitica species is divided into six biotypes, which differ in their ability to cause illness. The Y. enterocolitica incidence in Finland has been among the highest in the EU, but there has been little information on the occurrence of different Y. enterocolitica biotypes. enterocolitica Yersinia In this thesis Y. enterocolitica strains isolated from Finnish patients were characterised and the symptoms and sources of infections were analysed RESEARCH RESEARCH in a case-control study. The majority of clinical isolates of Y. enterocolitica were found to belong to biotype 1A, the status of which as a true pathogen is controversial. Furthermore, the study investigated the microbiological identification and molecular typing methods for Y. enterocolitica. The MLVA method was found to be appropriate for investigating foodborne outbreaks. in Finland This study adds to the understanding of epidemiology of Y. enterocolitica in Finland and emphasises the importance of correct identification of Yersinia strains in order to evaluate the clinical importance of the microbiological findings. National Institute for Health and Welfare P.O. Box 30 (Mannerheimintie 166) FI-00271 Helsinki, Finland 117 Telephone: 358 29 524 6000 117 117 2013 ISBN 978-952-302-064-1 www.thl.fi RESEARCH NRO 117 2014 Leila M. Sihvonen Clinical isolates of Yersinia enterocolitica in Finland Identification and Epidemiology ACADEMIC DISSERTATION To be presented with the permission of the Faculty of Agriculture and Forestry, University of Helsinki, for public examination in Auditorium 1041, Biocenter 2, Viikinkaari 5, on 17.01.2014, at 12 noon. -

International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5575–5599 DOI 10.1099/Ijsem.0.001485
International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5575–5599 DOI 10.1099/ijsem.0.001485 Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Mobolaji Adeolu,† Seema Alnajar,† Sohail Naushad and Radhey S. Gupta Correspondence Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario, Radhey S. Gupta L8N 3Z5, Canada [email protected] Understanding of the phylogeny and interrelationships of the genera within the order ‘Enterobacteriales’ has proven difficult using the 16S rRNA gene and other single-gene or limited multi-gene approaches. In this work, we have completed comprehensive comparative genomic analyses of the members of the order ‘Enterobacteriales’ which includes phylogenetic reconstructions based on 1548 core proteins, 53 ribosomal proteins and four multilocus sequence analysis proteins, as well as examining the overall genome similarity amongst the members of this order. The results of these analyses all support the existence of seven distinct monophyletic groups of genera within the order ‘Enterobacteriales’. In parallel, our analyses of protein sequences from the ‘Enterobacteriales’ genomes have identified numerous molecular characteristics in the forms of conserved signature insertions/deletions, which are specifically shared by the members of the identified clades and independently support their monophyly and distinctness. Many of these groupings, either in part or in whole, have been recognized in previous evolutionary studies, but have not been consistently resolved as monophyletic entities in 16S rRNA gene trees. The work presented here represents the first comprehensive, genome- scale taxonomic analysis of the entirety of the order ‘Enterobacteriales’. -

Add, Stir and Reduce' Mcnally, Alan; Thomson, Nicholas R; Reuter, Sandra; Wren, Brendan W
University of Birmingham 'Add, stir and reduce' McNally, Alan; Thomson, Nicholas R; Reuter, Sandra; Wren, Brendan W DOI: 10.1038/nrmicro.2015.29 License: Creative Commons: Attribution-NonCommercial-NoDerivs (CC BY-NC-ND) Document Version Peer reviewed version Citation for published version (Harvard): McNally, A, Thomson, NR, Reuter, S & Wren, BW 2016, ''Add, stir and reduce': Yersinia spp. as model bacteria for pathogen evolution', Nature Reviews Microbiology, vol. 14, no. 3, pp. 177-190. https://doi.org/10.1038/nrmicro.2015.29 Link to publication on Research at Birmingham portal Publisher Rights Statement: Checked 05/10/2016 General rights Unless a licence is specified above, all rights (including copyright and moral rights) in this document are retained by the authors and/or the copyright holders. The express permission of the copyright holder must be obtained for any use of this material other than for purposes permitted by law. •Users may freely distribute the URL that is used to identify this publication. •Users may download and/or print one copy of the publication from the University of Birmingham research portal for the purpose of private study or non-commercial research. •User may use extracts from the document in line with the concept of ‘fair dealing’ under the Copyright, Designs and Patents Act 1988 (?) •Users may not further distribute the material nor use it for the purposes of commercial gain. Where a licence is displayed above, please note the terms and conditions of the licence govern your use of this document. When citing, please reference the published version. Take down policy While the University of Birmingham exercises care and attention in making items available there are rare occasions when an item has been uploaded in error or has been deemed to be commercially or otherwise sensitive. -

Diagnostics and Epidemiology of Yersinia Pseudotuberculosis
DIAGNOSTICS AND EPIDEMIOLOGY OF YERSINIA PSEUDOTUBERCULOSIS Taina Niskanen Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki, Finland Helsinki 2010 Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki Helsinki, Finland DIAGNOSTICS AND EPIDEMIOLOGY OF YERSINIA PSEUDOTUBERCULOSIS Taina Niskanen ACADEMIC DISSERTATION To be presented with the permission of the Faculty of Veterinary Medicine, University of Helsinki, for public examination in Auditorium Arppeanum, Snellmaninkatu 3, Helsinki, on 29 January 2010, at 12 noon. Supervising Professor Professor Hannu Korkeala, DVM, PhD, MSocSc Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki Helsinki, Finland Supervised by Professor Hannu Korkeala, DVM, PhD, MSocSc Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki Helsinki, Finland Maria Fredriksson-Ahomaa, DVM, PhD Institute of Food Hygiene and Technology Faculty of Veterinary Medicine Ludwig-Maximilians-Universität Munich Germany Reviewed by Professor Dr. Lieven De Zutter Faculty of Veterinary Medicine Ghent University, Belgium Professor Stan Fenwick, BVMS, PhD Faculty of Health Sciences Murdoch University, Australia Opponent Docent Kaisa Granfors, PhD National Institute for Health and Welfare (THL) Turku, Finland ISBN 978-952-92-6589-3 (Paperback) ISBN 978-952-10-5921-6 (PDF) Helsinki University Print Helsinki 2009 Contents ACKNOWLEDGEMENTS ...........................................................................................................................6 -

WO 2019/094700 Al 16 May 2019 (16.05.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2019/094700 Al 16 May 2019 (16.05.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors: SANTOS, Michael; One Kendall Square, C07K 14/435 (2006.01) A01K 67/04 (2006.01) Building 200, Cambridge, Massachusetts 02139 (US). A01K 67/00 (2006.01) C07K 14/00 (2006.01) DELISLE, Scott; One Kendall Square, Building 200, Cam¬ A01K 67/033 (2006.01) bridge, Massachusetts 02139 (US). TWEED-KENT, Ailis; One Kendall Square, Building 200, Cambridge, Massa¬ (21) International Application Number: chusetts 02139 (US). EASTHON, Lindsey; One Kendall PCT/US20 18/059996 Square, Building 200, Cambridge, Massachusetts 02139 (22) International Filing Date: (US). PATTNI, Bhushan S.; 15 Evergreen Circle, Canton, 09 November 2018 (09. 11.2018) Massachusetts 02021 (US). (25) Filing Language: English (74) Agent: WARD, Donna T. et al; DT Ward, P.C., 142A Main Street, Groton, Massachusetts 01450 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/584,153 10 November 2017 (10. 11.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/659,213 18 April 2018 (18.04.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, 62/659,209 18 April 2018 (18.04.2018) US DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, 62/680,386 04 June 2018 (04.06.2018) US HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, 62/680,371 04 June 2018 (04.06.2018) US KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (71) Applicant: COCOON BIOTECH INC. -
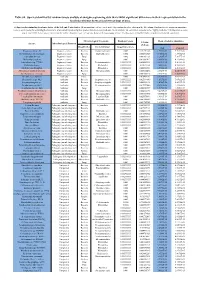
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp.