E. Coli (Expec) Among E
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Etude Et Identification De Yersinia Enterocolitica. Détermination Des Profils Antibiotypiques Et Électrophorétiques De L'adn Total
الجمهورٌــــــة الجزائرٌـــــــة الدٌمقراطٌـــــة الشعبٌــــــة République Algérienne Démocratique et Populaire وزارة التعلــٌم العالـــً والبحث العلمـــً Ministère de l’Enseignement Supérieure et la Recherche scientifique جامعة اﻹخوة منتوري قسنطٌنة Université des frères Mentouri Constantine Faculté des science de la nature et de la vie كلٌة علوم الطبٌعة والحٌاة Mémoire présenté en vue de l’obtention du Diplôme de Master Domaine : Science de la Nature et de la Vie Filière : Sciences Biologiques Spécialité : Microbiologie Générale et Biologie Moléculaire des Microorganismes Intitulé: Etude et identification de Yersinia enterocolitica. Détermination des profils antibiotypiques et électrophorétiques de l'ADN total Présenté par : BOUMESSRANE ROKIA Le : 24/06/2018 RABHI SAIDA Jury d’évaluation : Présidente : Mme RIAH. N (MCB-UFM Constantine1) Rapporteur : Mme BOUZERAIB. L (MAA-UFM Constantine1) Examinateur : Mr CHABBI.R (MAA-UFM Constantine1) Année universitaire 2017-2018 Remerciements C’est grâce à Allah le miséricordieux que l’aube du savoir à évacuer l’obscurité de l’ignorance et le soleil de la science à éclairer notre chemin pour réaliser ce modeste travail. Ce travail a été réalisé au sein de laboratoire de Zoologie de la faculté de science de la nature et de la vie université Mentouri Constantine 1. Nous adressons notre remerciement à madame BOUZERAIB LATIFA l’encadreur de notre mémoire : pour l’effort fourni, pour ses aides et gentillesse, pour les conseils qu’elle prodigués, sa patience et sa persévérance dans le suivi tout au long de la réalisation de ce travail, ainsi que pour sa bienveillance et ses qualités profondément humaines qui ont été remarquables. Nos remerciements ne sont jamais assez pour vous Madame. -

Epidemiology and Comparative Analysis of Yersinia in Ireland Author(S) Ringwood, Tamara Publication Date 2013 Original Citation Ringwood, T
UCC Library and UCC researchers have made this item openly available. Please let us know how this has helped you. Thanks! Title Epidemiology and comparative analysis of Yersinia in Ireland Author(s) Ringwood, Tamara Publication date 2013 Original citation Ringwood, T. 2013. Epidemiology and comparative analysis of Yersinia in Ireland. PhD Thesis, University College Cork. Type of publication Doctoral thesis Rights © 2013, Tamara Ringwood http://creativecommons.org/licenses/by-nc-nd/3.0/ Item downloaded http://hdl.handle.net/10468/1294 from Downloaded on 2021-10-07T12:07:10Z Epidemiology and Comparative Analysis of Yersinia in Ireland by Tamara Ringwood A thesis presented for the Degree of Doctor of Philosophy National University of Ireland, Cork University College Cork Coláiste na hOllscoile Corcaigh Department of Microbiology Head of Department: Prof. Gerald F. Fitzgerald Supervisor: Prof. Michael B. Prentice April 2013 Contents List of Tables .............................................................................................................................................. iv List of figures ............................................................................................................................................. vi Declaration ............................................................................................................................................... viii Acknowledgements ................................................................................................................................ -

Tierärztliche Hochschule Hannover Untersuchungen Zum Auftreten
Tierärztliche Hochschule Hannover Untersuchungen zum Auftreten verschiedener bakterieller Zoonoseerreger und zu den Risikofaktoren in norddeutschen Schweinemastbeständen INAUGURAL – DISSERTATION zur Erlangung des Grades einer Doktorin oder eines Doktors der Veterinärmedizin - Doctor medicinae veterinariae - ( Dr. med. vet. ) vorgelegt von Susanne Döhne Marburg/Lahn Hannover 2010 Wissenschaftliche Betreuung: Prof. Dr. L. Kreienbrock, Institut für Biometrie, Epidemiologie und Informationsverarbeitung Prof. Dr. K.-H. Waldmann, Klinik für kleine Klauentiere und forensische Medizin und Ambulatorische Klinik 1. Gutachter: Prof. Dr. L. Kreienbrock, Prof. Dr. K.-H. Waldmann 2. Gutachter: Prof. Dr. T. Blaha Tag der mündlichen Prüfung: 03.11.2010 Diese Dissertation wurde gefördert durch das Bundesministerium für Bildung und Forschung im Rahmen des Projektes FBI-Zoo Veröffentlichungen Erste Ergebnisse dieser Arbeit wurden bereits veröffentlicht: DÖHNE, S., A. VON ALTROCK, R. MERLE, K.-H. WALDMANN u. L. KREIENBROCK (2009): Incidence and antimicrobial susceptibility of Salmonella in fattening pig herds in Northern Germany. In: 14 th ISAH Congress Vechta, 19.-23. Juli 2009, Proceedings Vol.2 DÖHNE, S., A. VON ALTROCK, R. MERLE, K.-H. WALDMANN u. L. KREIENBROCK (2009): Resistenzen von Salmonella spp. in norddeutschen Schweinemastbetrieben. In: DVG-Fachgruppentagung Epidemiologie und Dokumentation: „Krankheitsdynamik in Populationen – Bedeutung von Surveillance und Impfprogrammen“, Gießen, 2.-4. September 2009 Inhaltsverzeichnis 1 Einleitung 13 2 Literaturübersicht -

Clinical Isolates of Yersinia Enterocolitica in Finland 117 2013 117
Leila M. Sihvonen Leila Leila M. Sihvonen Clinical isolates of Yersinia enterocolitica in Finland Leila M. Sihvonen Identification and Epidemiology Clinical isolates of Yersinia enterocolitica in Finland Identification and Epidemiology Clinical isolates of Clinical isolates Yersinia enterocolitica is a foodborne bacterium that causes gastroenteritis and post-infectious complications, such as reactive arthritis, in humans. Y. enterocolitica species is divided into six biotypes, which differ in their ability to cause illness. The Y. enterocolitica incidence in Finland has been among the highest in the EU, but there has been little information on the occurrence of different Y. enterocolitica biotypes. enterocolitica Yersinia In this thesis Y. enterocolitica strains isolated from Finnish patients were characterised and the symptoms and sources of infections were analysed RESEARCH RESEARCH in a case-control study. The majority of clinical isolates of Y. enterocolitica were found to belong to biotype 1A, the status of which as a true pathogen is controversial. Furthermore, the study investigated the microbiological identification and molecular typing methods for Y. enterocolitica. The MLVA method was found to be appropriate for investigating foodborne outbreaks. in Finland This study adds to the understanding of epidemiology of Y. enterocolitica in Finland and emphasises the importance of correct identification of Yersinia strains in order to evaluate the clinical importance of the microbiological findings. National Institute for Health and Welfare P.O. Box 30 (Mannerheimintie 166) FI-00271 Helsinki, Finland 117 Telephone: 358 29 524 6000 117 117 2013 ISBN 978-952-302-064-1 www.thl.fi RESEARCH NRO 117 2014 Leila M. Sihvonen Clinical isolates of Yersinia enterocolitica in Finland Identification and Epidemiology ACADEMIC DISSERTATION To be presented with the permission of the Faculty of Agriculture and Forestry, University of Helsinki, for public examination in Auditorium 1041, Biocenter 2, Viikinkaari 5, on 17.01.2014, at 12 noon. -

International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5575–5599 DOI 10.1099/Ijsem.0.001485
International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5575–5599 DOI 10.1099/ijsem.0.001485 Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Mobolaji Adeolu,† Seema Alnajar,† Sohail Naushad and Radhey S. Gupta Correspondence Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario, Radhey S. Gupta L8N 3Z5, Canada [email protected] Understanding of the phylogeny and interrelationships of the genera within the order ‘Enterobacteriales’ has proven difficult using the 16S rRNA gene and other single-gene or limited multi-gene approaches. In this work, we have completed comprehensive comparative genomic analyses of the members of the order ‘Enterobacteriales’ which includes phylogenetic reconstructions based on 1548 core proteins, 53 ribosomal proteins and four multilocus sequence analysis proteins, as well as examining the overall genome similarity amongst the members of this order. The results of these analyses all support the existence of seven distinct monophyletic groups of genera within the order ‘Enterobacteriales’. In parallel, our analyses of protein sequences from the ‘Enterobacteriales’ genomes have identified numerous molecular characteristics in the forms of conserved signature insertions/deletions, which are specifically shared by the members of the identified clades and independently support their monophyly and distinctness. Many of these groupings, either in part or in whole, have been recognized in previous evolutionary studies, but have not been consistently resolved as monophyletic entities in 16S rRNA gene trees. The work presented here represents the first comprehensive, genome- scale taxonomic analysis of the entirety of the order ‘Enterobacteriales’. -

T4-Like Bacteriophages Isolated from Pig Stools Infect Yersinia Pseudotuberculosis and Yersinia Pestis Using LPS and Ompf As Receptors
viruses Article T4-like Bacteriophages Isolated from Pig Stools Infect Yersinia pseudotuberculosis and Yersinia pestis Using LPS and OmpF as Receptors Mabruka Salem 1,2, Maria I. Pajunen 1 , Jin Woo Jun 3 and Mikael Skurnik 1,4,* 1 Department of Bacteriology and Immunology, Medicum, Human Microbiome Research Program, Faculty of Medicine, University of Helsinki, 00290 Helsinki, Finland; mabruka.salem@helsinki.fi (M.S.); maria.pajunen@helsinki.fi (M.I.P.) 2 Department of Microbiology, Faculty of Medicine, University of Benghazi, Benghazi 16063, Libya 3 Department of Aquaculture, Korea National College of Agriculture and Fisheries, Jeonju 54874, Korea; [email protected] 4 Division of Clinical Microbiology, Helsinki University Hospital, HUSLAB, 00290 Helsinki, Finland * Correspondence: mikael.skurnik@helsinki.fi; Tel.: +358-50-336-0981 Abstract: The Yersinia bacteriophages fPS-2, fPS-65, and fPS-90, isolated from pig stools, have long contractile tails and elongated heads, and they belong to genus Tequatroviruses in the order Caudovirales. The phages exhibited relatively wide host ranges among Yersinia pseudotuberculosis and related species. One-step growth curve experiments revealed that the phages have latent periods of 50–80 min with burst sizes of 44–65 virions per infected cell. The phage genomes consist of circularly permuted dsDNA of 169,060, 167,058, and 167,132 bp in size, respectively, with a G + C content 35.3%. The number of predicted genes range from 267 to 271. The phage genomes are 84–92% identical to each other and ca 85% identical to phage T4. The phage receptors were identified by whole genome Citation: Salem, M.; Pajunen, M.I.; Jun, J.W.; Skurnik, M. -

Add, Stir and Reduce' Mcnally, Alan; Thomson, Nicholas R; Reuter, Sandra; Wren, Brendan W
University of Birmingham 'Add, stir and reduce' McNally, Alan; Thomson, Nicholas R; Reuter, Sandra; Wren, Brendan W DOI: 10.1038/nrmicro.2015.29 License: Creative Commons: Attribution-NonCommercial-NoDerivs (CC BY-NC-ND) Document Version Peer reviewed version Citation for published version (Harvard): McNally, A, Thomson, NR, Reuter, S & Wren, BW 2016, ''Add, stir and reduce': Yersinia spp. as model bacteria for pathogen evolution', Nature Reviews Microbiology, vol. 14, no. 3, pp. 177-190. https://doi.org/10.1038/nrmicro.2015.29 Link to publication on Research at Birmingham portal Publisher Rights Statement: Checked 05/10/2016 General rights Unless a licence is specified above, all rights (including copyright and moral rights) in this document are retained by the authors and/or the copyright holders. The express permission of the copyright holder must be obtained for any use of this material other than for purposes permitted by law. •Users may freely distribute the URL that is used to identify this publication. •Users may download and/or print one copy of the publication from the University of Birmingham research portal for the purpose of private study or non-commercial research. •User may use extracts from the document in line with the concept of ‘fair dealing’ under the Copyright, Designs and Patents Act 1988 (?) •Users may not further distribute the material nor use it for the purposes of commercial gain. Where a licence is displayed above, please note the terms and conditions of the licence govern your use of this document. When citing, please reference the published version. Take down policy While the University of Birmingham exercises care and attention in making items available there are rare occasions when an item has been uploaded in error or has been deemed to be commercially or otherwise sensitive. -

The Quest for Novel Extracellular Polymers Produced by Soil-Borne Bacteria
Copyright is owned by the Author of the thesis. Permission is given for a copy to be downloaded by an individual for the purpose of research and private study only. The thesis may not be reproduced elsewhere without the permission of the Author. Bioprospecting: The quest for novel extracellular polymers produced by soil-borne bacteria A thesis presented in partial fulfilment of the requirements for the degree of Master of Science In Microbiology at Massey University, Palmerston North, New Zealand Jason Smith 2017 i Dedication This thesis is dedicated to my dad. Vaughan Peter Francis Smith 13 July 1955 – 27 April 2002 Though our time together was short you are never far from my mind nor my heart. ii Abstract Bacteria are ubiquitous in nature, and the surrounding environment. Bacterially produced extracellular polymers, and proteins are of particular value in the fields of medicine, food, science, and industry. Soil is an extremely rich source of bacteria with over 100 million per gram of soil, many of which produce extracellular polymers. Approximately 90% of soil-borne bacteria are yet to be cultured and classified. Here we employed an exploratory approach and culture based method for the isolation of soil-borne bacteria, and assessed their capability for extracellular polymer production. Bacteria that produced mucoid (of a mucous nature) colonies were selected for identification, imaging, and polymer production. Here we characterised three bacterial isolates that produced extracellular polymers, with a focus on one isolate that formed potentially novel proteinaceous cell surface appendages. These appendages have an unknown function, however, I suggest they may be important for bacterial communication, signalling, and nutrient transfer. -

Diagnostics and Epidemiology of Yersinia Pseudotuberculosis
DIAGNOSTICS AND EPIDEMIOLOGY OF YERSINIA PSEUDOTUBERCULOSIS Taina Niskanen Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki, Finland Helsinki 2010 Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki Helsinki, Finland DIAGNOSTICS AND EPIDEMIOLOGY OF YERSINIA PSEUDOTUBERCULOSIS Taina Niskanen ACADEMIC DISSERTATION To be presented with the permission of the Faculty of Veterinary Medicine, University of Helsinki, for public examination in Auditorium Arppeanum, Snellmaninkatu 3, Helsinki, on 29 January 2010, at 12 noon. Supervising Professor Professor Hannu Korkeala, DVM, PhD, MSocSc Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki Helsinki, Finland Supervised by Professor Hannu Korkeala, DVM, PhD, MSocSc Department of Food and Environmental Hygiene Faculty of Veterinary Medicine University of Helsinki Helsinki, Finland Maria Fredriksson-Ahomaa, DVM, PhD Institute of Food Hygiene and Technology Faculty of Veterinary Medicine Ludwig-Maximilians-Universität Munich Germany Reviewed by Professor Dr. Lieven De Zutter Faculty of Veterinary Medicine Ghent University, Belgium Professor Stan Fenwick, BVMS, PhD Faculty of Health Sciences Murdoch University, Australia Opponent Docent Kaisa Granfors, PhD National Institute for Health and Welfare (THL) Turku, Finland ISBN 978-952-92-6589-3 (Paperback) ISBN 978-952-10-5921-6 (PDF) Helsinki University Print Helsinki 2009 Contents ACKNOWLEDGEMENTS ...........................................................................................................................6 -

WO 2019/094700 Al 16 May 2019 (16.05.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2019/094700 Al 16 May 2019 (16.05.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors: SANTOS, Michael; One Kendall Square, C07K 14/435 (2006.01) A01K 67/04 (2006.01) Building 200, Cambridge, Massachusetts 02139 (US). A01K 67/00 (2006.01) C07K 14/00 (2006.01) DELISLE, Scott; One Kendall Square, Building 200, Cam¬ A01K 67/033 (2006.01) bridge, Massachusetts 02139 (US). TWEED-KENT, Ailis; One Kendall Square, Building 200, Cambridge, Massa¬ (21) International Application Number: chusetts 02139 (US). EASTHON, Lindsey; One Kendall PCT/US20 18/059996 Square, Building 200, Cambridge, Massachusetts 02139 (22) International Filing Date: (US). PATTNI, Bhushan S.; 15 Evergreen Circle, Canton, 09 November 2018 (09. 11.2018) Massachusetts 02021 (US). (25) Filing Language: English (74) Agent: WARD, Donna T. et al; DT Ward, P.C., 142A Main Street, Groton, Massachusetts 01450 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/584,153 10 November 2017 (10. 11.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/659,213 18 April 2018 (18.04.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, 62/659,209 18 April 2018 (18.04.2018) US DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, 62/680,386 04 June 2018 (04.06.2018) US HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, 62/680,371 04 June 2018 (04.06.2018) US KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (71) Applicant: COCOON BIOTECH INC. -
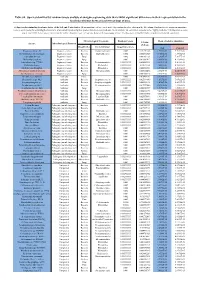
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Comparativa Genómica Y Expresión Diferencial De Genes En Respuesta a Temperatura Y Su Relación Con El Proceso Infeccioso De Yersinia Ruckeri
Programa de Doctorado Biología Funcional y Molecular Comparativa genómica y expresión diferencial de genes en respuesta a temperatura y su relación con el proceso infeccioso de Yersinia ruckeri Desirée Cascales Freire Tesis doctoral Oviedo 2017 2 4 6 7 8 Agradecimientos 9 10 11 12 Índice 13 14 Índice Índice ............................................................................................................................................................... 13 Abreviaturas y siglas .................................................................................................................................. 21 1. Introducción ........................................................................................................................................ 25 1.1. Situación de la acuicultura y sus principales patologías bacterianas asociadas ........ 27 1.2. Yersinia ruckeri..................................................................................................................................... 27 1.3. La enfermedad entérica de la boca roja (enteric redmouth disease) ............................. 29 1.4. Factores de virulencia de Y. ruckeri ............................................................................................. 33 1.5. Influencia de la temperatura en la expresión de genes de virulencia en bacterias patógenas de animales ectotermos .................................................................................................... 35 1.6. Efecto de la exposición a antibióticos en la virulencia