Rapid Separations of Protactinium and Uranium Radioisotopes From
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Table 2.Iii.1. Fissionable Isotopes1
FISSIONABLE ISOTOPES Charles P. Blair Last revised: 2012 “While several isotopes are theoretically fissionable, RANNSAD defines fissionable isotopes as either uranium-233 or 235; plutonium 238, 239, 240, 241, or 242, or Americium-241. See, Ackerman, Asal, Bale, Blair and Rethemeyer, Anatomizing Radiological and Nuclear Non-State Adversaries: Identifying the Adversary, p. 99-101, footnote #10, TABLE 2.III.1. FISSIONABLE ISOTOPES1 Isotope Availability Possible Fission Bare Critical Weapon-types mass2 Uranium-233 MEDIUM: DOE reportedly stores Gun-type or implosion-type 15 kg more than one metric ton of U- 233.3 Uranium-235 HIGH: As of 2007, 1700 metric Gun-type or implosion-type 50 kg tons of HEU existed globally, in both civilian and military stocks.4 Plutonium- HIGH: A separated global stock of Implosion 10 kg 238 plutonium, both civilian and military, of over 500 tons.5 Implosion 10 kg Plutonium- Produced in military and civilian 239 reactor fuels. Typically, reactor Plutonium- grade plutonium (RGP) consists Implosion 40 kg 240 of roughly 60 percent plutonium- Plutonium- 239, 25 percent plutonium-240, Implosion 10-13 kg nine percent plutonium-241, five 241 percent plutonium-242 and one Plutonium- percent plutonium-2386 (these Implosion 89 -100 kg 242 percentages are influenced by how long the fuel is irradiated in the reactor).7 1 This table is drawn, in part, from Charles P. Blair, “Jihadists and Nuclear Weapons,” in Gary A. Ackerman and Jeremy Tamsett, ed., Jihadists and Weapons of Mass Destruction: A Growing Threat (New York: Taylor and Francis, 2009), pp. 196-197. See also, David Albright N 2 “Bare critical mass” refers to the absence of an initiator or a reflector. -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -
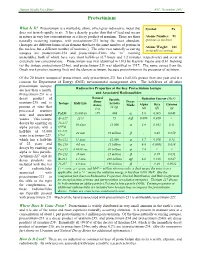
Protactinium
Human Health Fact Sheet ANL, November 2001 Protactinium What Is It? Protactinium is a malleable, shiny, silver-gray radioactive metal that Symbol: Pa does not tarnish rapidly in air. It has a density greater than that of lead and occurs in nature in very low concentrations as a decay product of uranium. There are three Atomic Number: 91 naturally occurring isotopes, with protactinium-231 being the most abundant. (protons in nucleus) (Isotopes are different forms of an element that have the same number of protons in Atomic Weight: 231 the nucleus but a different number of neutrons.) The other two naturally occurring (naturally occurring) isotopes are protactinium-234 and protactinium-234m (the “m” meaning metastable), both of which have very short half-lives (6.7 hours and 1.2 minutes, respectively) and occur in extremely low concentrations. Protactinium was first identified in 1913 by Kasimir Fajans and O.H. Gohring (as the isotope protactinium-234m), and protactinium-231 was identified in 1917. The name comes from the Greek work protos (meaning first) and the element actinium, because protactinium is the precursor of actinium. Of the 20 known isotopes of protactinium, only protactinium-231 has a half-life greater than one year and is a concern for Department of Energy (DOE) environmental management sites. The half-lives of all other protactinium isotopes Radioactive Properties of the Key Protactinium Isotope are less than a month. Protactinium-231 is a and Associated Radionuclides Natural decay product of Specific Radiation Energy (MeV) Abun- Decay uranium-235 and is Isotope Half-Life Activity dance Mode Alpha Beta Gamma present at sites that (Ci/g) (%) (α) (β) (γ) processed uranium α ores and associated Pa231 33,000 yr >99 .048 5.0 0.065 0.048 wastes. -

A Global Scavenging and Circulation Ocean Model of Thorium-230 and Protactinium-231 with Improved Particle Dynamics (NEMO–Prothorp 0.1)
Geosci. Model Dev., 11, 3537–3556, 2018 https://doi.org/10.5194/gmd-11-3537-2018 © Author(s) 2018. This work is distributed under the Creative Commons Attribution 4.0 License. A global scavenging and circulation ocean model of thorium-230 and protactinium-231 with improved particle dynamics (NEMO–ProThorP 0.1) Marco van Hulten1,2, Jean-Claude Dutay1, and Matthieu Roy-Barman1 1Laboratoire des Sciences du Climat et de l’Environnement, IPSL, CEA–Orme des Merisiers, 91191 Gif-sur-Yvette, France 2Geophysical Institute, University of Bergen, Bergen, Norway Correspondence: Marco van Hulten ([email protected]) Received: 1 November 2017 – Discussion started: 8 December 2017 Revised: 16 August 2018 – Accepted: 20 August 2018 – Published: 31 August 2018 Abstract. In this paper we set forth a 3-D ocean model of the can be done based on our model as its source code is readily radioactive trace isotopes 230Th and 231Pa. The interest arises available. from the fact that these isotopes are extensively used for in- vestigating particle transport in the ocean and reconstructing past ocean circulation. The tracers are reversibly scavenged 1 Introduction by biogenic and lithogenic particles. Our simulations of 230Th and 231Pa are based on the Oceanic circulation and the carbon cycle play a major role NEMO–PISCES ocean biogeochemistry general circulation in the regulation of the past and present climate. Heat and model, which includes biogenic particles, namely small and carbon dioxide in the atmosphere tend to equilibrate with the big particulate organic carbon, calcium carbonate and bio- ocean surface and are transported down into the deep ocean genic silica. -

Upper Limit of the Periodic Table and the Future Superheavy Elements
CLASSROOM Rajarshi Ghosh Upper Limit of the Periodic Table and the Future Department of Chemistry The University of Burdwan ∗ Superheavy Elements Burdwan 713 104, India. Email: [email protected] Controversy surrounds the isolation and stability of the fu- ture transactinoid elements (after oganesson) in the periodic table. A single conclusion has not yet been drawn for the highest possible atomic number, though there are several the- oretical as well as experimental results regarding this. In this article, the scientific backgrounds of those upcoming super- heavy elements (SHE) and their proposed electronic charac- ters are briefly described. Introduction Totally 118 elements, starting from hydrogen (atomic number 1) to oganesson (atomic number 118) are accommodated in the mod- ern form of the periodic table comprising seven periods and eigh- teen groups. Total 92 natural elements (if technetium is consid- ered as natural) are there in the periodic table (up to uranium hav- ing atomic number 92). In the actinoid series, only four elements— Keywords actinium, thorium, protactinium and uranium—are natural. The Superheavy elements, actinoid rest of the eleven elements—from neptunium (atomic number 93) series, transactinoid elements, periodic table. to lawrencium (atomic number 103)—are synthetic. Elements after actinoids (i.e., from rutherfordium) are called transactinoid elements. These are also called superheavy elements (SHE) as they have very high atomic numbers. Prof. G T Seaborg had Elements after actinoids a very distinct contribution in the field of transuranium element (i.e., from synthesis. For this, Prof. Seaborg was awarded the Nobel Prize in rutherfordium) are called transactinoid elements. 1951. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 2) [5/10/2001 3:08:31 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

S D F P Periodic Table of the Elements O
shells (energy levels) (energy shells Periodic Table of the Elements levels) (energy shells I A s d p f subshell subshell subshell subshell Note: 1+ Atomic number = # of protons = # of electrons VIII A Atomic mass (rounded to nearest integer) = # of protons + # of neutrons hydrogen KEY most common helium 1 2- oxidation number 2 1 II A 1 H oxygen element name III A IV A V A VI A VII A He 1.00794(7) atomic number 8 4.002602(2) 2+ 3+ 3- 2- 1- symbol lithium beryllium O boron carbon nitrogen oxygen fluorine neon 3 4 5 6 7 8 9 10 2 Li Be 2001 atomic mass 15.9994(3) B C N O F Ne 2 6.941(2) 9.012182(3) Note: The last significant figure is reliable to 4 orbital 10.811(7) 12.0107(8) 14.00674(7) 15.9994(3) 18.9984032(5) 20.1797(6) sodium magnesium +-1 except where greater uncertainty is given p aluminium silicon phosphorus sulfur chlorine argon 11 12 in parentheses ( ). Mass numbers given in 13 3+ 14 15 16 17 18 3 brackets [ ] are of the longest lived isotopes. 3 Na Mg III B IV B V B VI B VII B VIII B I B II B Al Si P S Cl Ar 22.989770(2) 24.3050(6) 26.981538(2) 28.0855(3) 30.973761(2) 32.065(5) 35.453(2) 39.948(1) potassium calcium scandium titanium vanadium chromium manganese iron cobalt nickel copper zinc gallium germanium arsenic selenium bromine krypton 19 20 21 22 23 24 25 26 27 28 29 30 2+ 31 3+ 32 33 34 35 36 4 K Ca 3 Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 4 39.0983(1) 40.078(4) 44.955910(8) 47.867(1) 50.9415(1) 51.9961(6) 54.938049(9) 55.845(2) 58.933200(9) 58.6934(2) 63.546(3) 65.409(4) 69.723(1) 72.64(1) 74.92160(2) 78.96(3) -

Sorption of Cesium, Radium, Protactinium, Uranium, Neptunium and Plutonium on Rapakivi Granite
Sorption of cesium, radium, protactinium, uranium, neptunium and plutonium on rapakivi granite Tuula Huitti, Martti Hakanen Laboratory of Radiochemistry Department of Chemistry University of Helsinki Antero Lindberg Geological Survey of Finland December 1996 POSIVA OY Annankatu 42 D, FIN-00100 HELSINKI. FINLAND Phone (09) 2280 30 (nat.), (+358-9-) 2280 30 (int.) Fax (09) 2280 3719 (nat.). (+358-9-) 2280 3719 (int.) ISBN 951-652-022-7 ISSN 1239-3096 The conclusions and viewpoints presented in the report ere those of author(s) and do not necessarily coincide with those of Posiva. i - POSiVa report Raportmtunnus-Reponcode POSIVA-96-23 Annankatu 42 D, FIN-00100 HELSINKI, FINLAND Juikasuaika-Daie Puh. (09) 2280 30 - Int. Tel. +358 9 2280 30 December 1996 Tekija(t) - Author(s) Toimeksiantaja(t) - Commissioned by Tuula Huitti, Martti Hakanen, University of Helsinki Posiva Oy Antero Lindberg, Geological Survey of Finland Nimeke - Title SORPTION OF CESIUM, RADIUM, PROTACTINIUM, URANIUM, NEPTUNIUM AND PLUTONIUM ON RAPAKIVI GRANITE Thvistelma - Abstract Study was made of the sorption properties of rapakivi granite at Hasholmen in Loviisa for nuclides of spent fuel. R<j values of cesium, radium, protactinium, uranium, neptunium and plutonium were determined and diffusion of water in the rapakivi was investigated. The rock samples were taken by core drilling from the wall of the repository for operating waste and represented three different alteration stages: fresh, weathered and altered. The rock was crushed to grain size < 2 mm. Water used in the experiments was a brackish groundwater from Hastholmen. The rock material was characterized by determination of cation exchange capacities, specific areas (N2/BET) and volumetric porosities. -

Group 1: the Alkali Metal Family
Group 1: The Alkali Metal Family Location: The alkali metal family is found on the periodic table in Group 1, which is on the far left side of the table. The metals in this group are lithium, sodium, potassium, rubidium, cesium, and francium. The gas hydrogen is also put in this group because it shares similar reactivity with the alkali metals. Properties: All the metals in this family are silvery-white and soft enough to cut with a knife. They have low melting points. These metals, along with hydrogen, are extremely reactive. Hydrogen will blow up if it comes into any contact with flames. These metals are so reactive they will burn the skin if touched, so they are not safe for humans to handle. They tarnish rapidly. The metals in this family react violently with water. They easily react to form salts with elements from the halogen family. Because these elements are so reactive, they are never found in their pure forms in nature. In nature, they are always found combined with other elements. The metals in this family are easy to identify because they each give off a different color when they burn. Lithium flames are a crimson color, sodium flames are yellow, potassium flames are violet, rubidium flames are reddish-violet, and cesium flames are blue. Little is known about francium because it is so rare and it is radioactive. Uses: The alkali metal family has many important uses. Lithium is used in grease and other lubricants. It is also used in aircraft parts and batteries. Sodium is found in salt, and used in gasoline. -

PROTACTINIUM Element Symbol: Pa Atomic Number: 91
PROTACTINIUM Element Symbol: Pa Atomic Number: 91 An initiative of IYC 2011 brought to you by the RACI FRED DUNCAN www.raci.org.au PROTACTINIUM Element symbol: Pa Atomic number: 91 Protactinium has the symbol Pa, and is the ninety first element of the periodic table. In a pure form, it would be a dense, silver-grey metal – but it is extremely rare with an average concentration in the Earth’s crust of a few parts per trillion. There are several known isotopes of Protactinium, all of which are intrinsically unstable and undergo radioactive decay with half lives ranging from minutes up to many thousands of years. The most common isotope (with 99.9% natural abundance) is Pa-231, a member of the Uranium-235 decay series with a half life of 32700 years, while other less common forms are found as members of Thorium and U-238 decay chains. The element does not exist in nature except as a member of decay series’, and has no known uses outside of scientific research although it is used as a marker in the radiometric dating of sediments. In 1961 the UK Atomic Energy Agency extracted 125g of Protactinium from 60 tonnes of spent reactor fuel at a cost of half a million $US, which for many years comprised the entire known world stocks of the pure element. More recently, Oak Ridge National Laboratory in the US has been producing Pa for research purposes at a cost of approximately $280/gram. Protactinium-231 decays by the emission of an alpha particle to Actinium – other isotopes (which exist only in trace amounts) decay by the emission of a beta particle.