The Crystal Structure of Myelin Oligodendrocyte Glycoprotein, a Key Autoantigen in Multiple Sclerosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Myelin Biogenesis Is Associated with Pathological Ultrastructure That Is
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Myelin biogenesis is associated with pathological ultrastructure that 2 is resolved by microglia during development 3 4 5 Minou Djannatian1,2*, Ulrich Weikert3, Shima Safaiyan1,2, Christoph Wrede4, Cassandra 6 Deichsel1,2, Georg Kislinger1,2, Torben Ruhwedel3, Douglas S. Campbell5, Tjakko van Ham6, 7 Bettina Schmid2, Jan Hegermann4, Wiebke Möbius3, Martina Schifferer2,7, Mikael Simons1,2,7* 8 9 1Institute of Neuronal Cell Biology, Technical University Munich, 80802 Munich, Germany 10 2German Center for Neurodegenerative Diseases (DZNE), 81377 Munich, Germany 11 3Max-Planck Institute of Experimental Medicine, 37075 Göttingen, Germany 12 4Institute of Functional and Applied Anatomy, Research Core Unit Electron Microscopy, 13 Hannover Medical School, 30625, Hannover, Germany 14 5Department of Neuronal Remodeling, Graduate School of Pharmaceutical Sciences, Kyoto 15 University, Sakyo-ku, Kyoto 606-8501, Japan. 16 6Department of Clinical Genetics, Erasmus MC, University Medical Center Rotterdam, 3015 CN, 17 the Netherlands 18 7Munich Cluster of Systems Neurology (SyNergy), 81377 Munich, Germany 19 20 *Correspondence: [email protected] or [email protected] 21 Keywords 22 Myelination, degeneration, phagocytosis, microglia, oligodendrocytes, phosphatidylserine 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
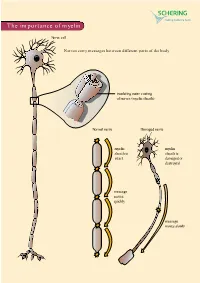
The Importance of Myelin
The importance of myelin Nerve cell Nerves carry messages between different parts of the body insulating outer coating of nerves (myelin sheath) Normal nerve Damaged nerve myelin myelin sheath is sheath is intact damaged or destroyed message moves quickly message moves slowly Nerve cells transmit impulses Nerve cells have a long, thin, flexible fibre that transmits impulses. These impulses are electrical signals that travel along the length of the nerve. Nerve fibres are long to enable impulses to travel between distant parts of the body, such as the spinal cord and leg muscles. Myelin speeds up impulses Most nerve fibres are surrounded by an insulating, fatty sheath called myelin, which acts to speed up impulses. The myelin sheath contains periodic breaks called nodes of Ranvier. By jumping from node to node, the impulse can travel much more quickly than if it had to travel along the entire length of the nerve fibre. Myelinated nerves can transmit a signal at speeds as high as 100 metres per second – as fast as a Formula One racing car. normal damaged nerve nerve Loss of myelin leads to a variety of symptoms If the myelin sheath surrounding nerve fibres is damaged or destroyed, transmission of nerve impulses is slowed or blocked. The impulse now has to flow continuously along the whole nerve fibre – a process that is much slower than jumping from node to node. Loss of myelin can also lead to ‘short-circuiting’ of nerve impulses. An area where myelin has been destroyed is called a lesion or plaque. This slowing and ‘short-circuiting’ of nerve impulses by lesions leads to a variety of symptoms related to nervous system activity. -

Regulation of Myelin Structure and Conduction Velocity by Perinodal Astrocytes
Correction NEUROSCIENCE Correction for “Regulation of myelin structure and conduc- tion velocity by perinodal astrocytes,” by Dipankar J. Dutta, Dong Ho Woo, Philip R. Lee, Sinisa Pajevic, Olena Bukalo, William C. Huffman, Hiroaki Wake, Peter J. Basser, Shahriar SheikhBahaei, Vanja Lazarevic, Jeffrey C. Smith, and R. Douglas Fields, which was first published October 29, 2018; 10.1073/ pnas.1811013115 (Proc. Natl. Acad. Sci. U.S.A. 115,11832–11837). The authors note that the following statement should be added to the Acknowledgments: “We acknowledge Dr. Hae Ung Lee for preliminary experiments that informed the ultimate experimental approach.” Published under the PNAS license. Published online June 10, 2019. www.pnas.org/cgi/doi/10.1073/pnas.1908361116 12574 | PNAS | June 18, 2019 | vol. 116 | no. 25 www.pnas.org Downloaded by guest on October 2, 2021 Regulation of myelin structure and conduction velocity by perinodal astrocytes Dipankar J. Duttaa,b, Dong Ho Wooa, Philip R. Leea, Sinisa Pajevicc, Olena Bukaloa, William C. Huffmana, Hiroaki Wakea, Peter J. Basserd, Shahriar SheikhBahaeie, Vanja Lazarevicf, Jeffrey C. Smithe, and R. Douglas Fieldsa,1 aSection on Nervous System Development and Plasticity, The Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892; bThe Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., Bethesda, MD 20817; cMathematical and Statistical Computing Laboratory, Office of Intramural Research, Center for Information -
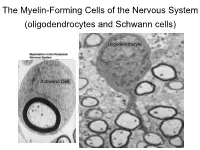
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Disease
MOG ANTIBODY DISEASE Myelin Oligodendrocyte Glycoprotein (MOG) Antibody-Associated Encephalomyelitis WHAT IS MOG ANTIBODY-ASSOCIATED DEMYELINATION? Myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination is an immune-mediated inflammatory process that affects the central nervous system (brain and spinal cord). MOG is a protein that exists on the outer surface of cells that create myelin (an insulating layer around nerve fibers). In a small number of patients, an initial episode of inflammation due to MOG antibodies may be the first manifestation of multiple sclerosis (MS). Most patients may only experience one episode of inflammation, but repeated episodes of central nervous system demyelination can occur in some cases. WHAT ARE THE SYMPTOMS? • Optic neuritis (inflammation of the optic nerve(s)) may be a symptom of MOG antibody- associated demyelination, which may result in painful loss of vision in one or both eyes. • Transverse myelitis (inflammation of the spinal cord) may cause a variety of symptoms that include: o Abnormal sensations. Numbness, tingling, coldness or burning in the arms and/or legs. Some are especially sensitive to the light touch of clothing or to extreme heat or cold. You may feel as if something is tightly wrapping the skin of your chest, abdomen or legs. o Weakness in your arms or legs. Some people notice that they're stumbling or dragging one foot, or heaviness in the legs. Others may develop severe weakness or even total paralysis. o Bladder and bowel problems. This may include needing to urinate more frequently, urinary incontinence, difficulty urinating and constipation. • Acute disseminated encephalomyelitis (ADEM) may cause loss of vision, weakness, numbness, and loss of balance, and altered mental status. -

Targeting Myelin Lipid Metabolism As a Potential Therapeutic Strategy in a Model of CMT1A Neuropathy
ARTICLE DOI: 10.1038/s41467-018-05420-0 OPEN Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy R. Fledrich 1,2,3, T. Abdelaal 1,4,5, L. Rasch1,4, V. Bansal6, V. Schütza1,3, B. Brügger7, C. Lüchtenborg7, T. Prukop1,4,8, J. Stenzel1,4, R.U. Rahman6, D. Hermes 1,4, D. Ewers 1,4, W. Möbius 1,9, T. Ruhwedel1, I. Katona 10, J. Weis10, D. Klein11, R. Martini11, W. Brück12, W.C. Müller3, S. Bonn 6,13, I. Bechmann2, K.A. Nave1, R.M. Stassart 1,3,12 & M.W. Sereda1,4 1234567890():,; In patients with Charcot–Marie–Tooth disease 1A (CMT1A), peripheral nerves display aberrant myelination during postnatal development, followed by slowly progressive demye- lination and axonal loss during adult life. Here, we show that myelinating Schwann cells in a rat model of CMT1A exhibit a developmental defect that includes reduced transcription of genes required for myelin lipid biosynthesis. Consequently, lipid incorporation into myelin is reduced, leading to an overall distorted stoichiometry of myelin proteins and lipids with ultrastructural changes of the myelin sheath. Substitution of phosphatidylcholine and phosphatidylethanolamine in the diet is sufficient to overcome the myelination deficit of affected Schwann cells in vivo. This treatment rescues the number of myelinated axons in the peripheral nerves of the CMT rats and leads to a marked amelioration of neuropathic symptoms. We propose that lipid supplementation is an easily translatable potential therapeutic approach in CMT1A and possibly other dysmyelinating neuropathies. 1 Department of Neurogenetics, Max-Planck-Institute of Experimental Medicine, Göttingen 37075, Germany. -

The Effects of Normal Aging on Myelin and Nerve Fibers: a Review∗
Journal of Neurocytology 31, 581–593 (2002) The effects of normal aging on myelin and nerve fibers: A review∗ ALAN PETERS Department of Anatomy and Neurobiology, Boston University School of Medicine, 715, Albany Street, Boston, MA 02118, USA [email protected] Received 7 January 2003; revised 4 March 2003; accepted 5 March 2003 Abstract It was believed that the cause of the cognitive decline exhibited by human and non-human primates during normal aging was a loss of cortical neurons. It is now known that significant numbers of cortical neurons are not lost and other bases for the cognitive decline have been sought. One contributing factor may be changes in nerve fibers. With age some myelin sheaths exhibit degenerative changes, such as the formation of splits containing electron dense cytoplasm, and the formation on myelin balloons. It is suggested that such degenerative changes lead to cognitive decline because they cause changes in conduction velocity, resulting in a disruption of the normal timing in neuronal circuits. Yet as degeneration occurs, other changes, such as the formation of redundant myelin and increasing thickness suggest of sheaths, suggest some myelin formation is continuing during aging. Another indication of this is that oligodendrocytes increase in number with age. In addition to the myelin changes, stereological studies have shown a loss of nerve fibers from the white matter of the cerebral hemispheres of humans, while other studies have shown a loss of nerve fibers from the optic nerves and anterior commissure in monkeys. It is likely that such nerve fiber loss also contributes to cognitive decline, because of the consequent decrease in connections between neurons. -

Schwann Cells, but Not Oligodendrocytes, Depend Strictly
RESEARCH ARTICLE Schwann cells, but not Oligodendrocytes, Depend Strictly on Dynamin 2 Function Daniel Gerber1†, Monica Ghidinelli1†, Elisa Tinelli1, Christian Somandin1, Joanne Gerber1, Jorge A Pereira1, Andrea Ommer1, Gianluca Figlia1, Michaela Miehe1, Lukas G Na¨ geli1, Vanessa Suter1, Valentina Tadini1, Pa´ ris NM Sidiropoulos1, Carsten Wessig2, Klaus V Toyka2, Ueli Suter1* 1Department of Biology, Institute of Molecular Health Sciences, Swiss Federal Institute of Technology, ETH Zurich, Zurich, Switzerland; 2Department of Neurology, University Hospital of Wu¨ rzburg, University of Wu¨ rzburg, Wu¨ rzburg, Germany Abstract Myelination requires extensive plasma membrane rearrangements, implying that molecules controlling membrane dynamics play prominent roles. The large GTPase dynamin 2 (DNM2) is a well-known regulator of membrane remodeling, membrane fission, and vesicular trafficking. Here, we genetically ablated Dnm2 in Schwann cells (SCs) and in oligodendrocytes of mice. Dnm2 deletion in developing SCs resulted in severely impaired axonal sorting and myelination onset. Induced Dnm2 deletion in adult SCs caused a rapidly-developing peripheral neuropathy with abundant demyelination. In both experimental settings, mutant SCs underwent prominent cell death, at least partially due to cytokinesis failure. Strikingly, when Dnm2 was deleted in adult SCs, non-recombined SCs still expressing DNM2 were able to remyelinate fast and efficiently, accompanied by neuropathy remission. These findings reveal a remarkable self-healing capability of peripheral nerves that are affected by SC loss. In the central nervous system, however, *For correspondence: we found no major defects upon Dnm2 deletion in oligodendrocytes. [email protected] DOI: https://doi.org/10.7554/eLife.42404.001 †These authors contributed equally to this work Competing interests: The Introduction authors declare that no Motor, sensory, and cognitive functions of the nervous system require rapid and refined impulse competing interests exist. -

Clinical Syndromes Associated with Tomacula Or Myelin Swellings in Sural Nerve Biopsies
J Neurol Neurosurg Psychiatry 2000;68:483–488 483 J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.68.4.483 on 1 April 2000. Downloaded from Clinical syndromes associated with tomacula or myelin swellings in sural nerve biopsies S Sander, R A Ouvrier, J G McLeod, G A Nicholson, J D Pollard Abstract syndromes.8–11 Sausage shaped swellings of the Objectives—To describe the neuropatho- myelin sheath were first described by Behse and logical features of clinical syndromes Buchthal in 1972.1 Madrid and Bradley10 subse- associated with tomacula or focal myelin quently gave the name tomaculous neuropathy swellings in sural nerve biospies and to (latin: tomaculum=sausage) and described sev- discuss possible common aetiopathologi- eral mechanisms that may lead to the formation cal pathways leading to their formation in of a tomaculum—for example, hypermyelina- this group of neuropathies. tion, redundant loop formation, the presence of Methods—Fifty two patients with sural a second mesaxon, transnodal myelination, two nerve biopsies reported to show tomacula Schwann cells forming one myelin sheath, and or focal myelin swellings were reviewed, disruption of the myelin sheath. Sural nerve light and electron microscopy were per- biopsies typically show regions of myelin thick- formed, and tomacula were analysed on ening as well as features of demyelination and teased fibre studies. Molecular genetic remyelination. Electrophysiologically, these syn- studies were performed on those patients dromes most often present as multiple monone- who were available for genetic testing. uropathy (sometimes with conduction block) or Results—Thirty seven patients were diag- demyelinating sensorimotor neuropathy. nosed with hereditary neuropathy with In this study we reviewed 52 patients show- liability to pressure palsies (HNPP), four ing myelin swellings on sural nerve biopsy. -

Normal Myelination a Practical Pictorial Review
Normal Myelination A Practical Pictorial Review Helen M. Branson, BSc, MBBS, FRACR KEYWORDS Myelin Myelination T1 T2 MR Diffusion KEY POINTS MR imaging is the best noninvasive modality to assess myelin maturation in the human brain. A combination of conventional T1-weighted and T2-weighted sequences is all that is required for basic assessment of myelination in the central nervous system (CNS). It is vital to have an understanding of the normal progression of myelination on MR imaging to enable the diagnosis of childhood diseases including leukodystrophies as well as hypomyelinating disorders, delayed myelination, and acquired demyelinating disease. INTRODUCTION and its role in the human nervous system is needed. Assessment of the progression of myelin and mye- Myelin is present in both the CNS and the lination has been revolutionized in the era of MR peripheral nervous system. In the CNS, it is imaging. Earlier imaging modalities such as ultra- primarily found in white matter (although small sonography and computed tomography have no amounts are also found in gray matter) and thus current role or ability to contribute to the as- is responsible for its color.1 Myelin acts as an elec- sessment of myelin maturation or abnormalities trical insulator for neurons.1 Myelin plays a role in of myelin. The degree of brain myelination can be increasing the speed of an action potential by used as a marker of maturation. 10–100 times that of an unmyelinated axon1 and The authors discuss also helps in speedy axonal transport.2 Edgar 3 1. Myelin function and structure and Garbern (2004) demonstrated that the ab- 2. -

Autophagy in Myelinating Glia
256 • The Journal of Neuroscience, January 8, 2020 • 40(2):256–266 Viewpoints Autophagy in Myelinating Glia Jillian Belgrad,1 XRaffaella De Pace,2 and R. Douglas Fields1 1Section on Nervous System Development and Plasticity and 2Section on Intracellular Protein Trafficking, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland 20892 Autophagy is the cellular process involved in transportation and degradation of membrane, proteins, pathogens, and organelles. This fundamental cellular process is vital in development, plasticity, and response to disease and injury. Compared with neurons, little informationisavailableonautophagyinglia,butitisparamountforgliatoperformtheircriticalresponsestonervoussystemdiseaseand injury, including active tissue remodeling and phagocytosis. In myelinating glia, autophagy has expanded roles, particularly in phago- cytosis of mature myelin and in generating the vast amounts of membrane proteins and lipids that must be transported to form new myelin. Notably, autophagy plays important roles in removing excess cytoplasm to promote myelin compaction and development of oligodendrocytes, as well as in remyelination by Schwann cells after nerve trauma. This review summarizes the cell biology of autophagy, detailing the major pathways and proteins involved, as well as the roles of autophagy in Schwann cells and oligodendrocytes in develop- ment, plasticity, and diseases in which myelin is affected. This includes traumatic brain injury, Alexander’s -

Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights Into the Disease Pathophysiology, Diagnosis and Management
International Journal of Molecular Sciences Review Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management Wojciech Ambrosius 1,*, Sławomir Michalak 2, Wojciech Kozubski 1 and Alicja Kalinowska 2 1 Department of Neurology, Poznan University of Medical Sciences, 49 Przybyszewskiego Street, 60-355 Poznan, Poland; [email protected] 2 Department of Neurology, Division of Neurochemistry and Neuropathology, Poznan University of Medical Sciences, 49 Przybyszewskiego Street, 60-355 Poznan, Poland; [email protected] (S.M.); [email protected] (A.K.) * Correspondence: [email protected]; Tel.: +48-61-869-1535 Abstract: Myelin oligodendrocyte glycoprotein (MOG)-associated disease (MOGAD) is a rare, antibody-mediated inflammatory demyelinating disorder of the central nervous system (CNS) with various phenotypes starting from optic neuritis, via transverse myelitis to acute demyelinating encephalomyelitis (ADEM) and cortical encephalitis. Even though sometimes the clinical picture of this condition is similar to the presentation of neuromyelitis optica spectrum disorder (NMOSD), most experts consider MOGAD as a distinct entity with different immune system pathology. MOG is a molecule detected on the outer membrane of myelin sheaths and expressed primarily within the brain, spinal cord and also the optic nerves. Its function is not fully understood but this glycoprotein may act as a cell surface receptor or cell adhesion molecule. The specific outmost location of myelin makes it a potential target for autoimmune antibodies and cell-mediated responses in demyelinating processes. Optic neuritis seems to be the most frequent presenting phenotype in adults and ADEM in children. In adults, the disease course is multiphasic and subsequent relapses increase disability.