Bcl11b Sets Pro-T Cell Fate by Site-Specific Cofactor Recruitment and by Repressing Id2 and Zbtb16
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 RESEARCH ARTICLE Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition Alexandre Puissant1,3, Stacey M. Frumm1,3, Gabriela Alexe1,3,5,6, Christopher F. Bassil1,3, Jun Qi2, Yvan H. Chanthery8, Erin A. Nekritz8, Rhamy Zeid2, William Clay Gustafson8, Patricia Greninger7, Matthew J. Garnett10, Ultan McDermott10, Cyril H. Benes7, Andrew L. Kung1,3, William A. Weiss8,9, James E. Bradner2,4, and Kimberly Stegmaier1,3,6 Downloaded from cancerdiscovery.aacrjournals.org on October 2, 2021. © 2013 American Association for Cancer Research. 15-CD-12-0418_p308-323.indd 1 22/02/13 12:15 AM Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 A BSTRACT Bromodomain inhibition comprises a promising therapeutic strategy in cancer, particularly for hematologic malignancies. To date, however, genomic biomarkers to direct clinical translation have been lacking. We conducted a cell-based screen of genetically defined cancer cell lines using a prototypical inhibitor of BET bromodomains. Integration of genetic features with chemosensitivity data revealed a robust correlation between MYCN amplification and sensitivity to bromodomain inhibition. We characterized the mechanistic and translational significance of this finding in neuroblastoma, a childhood cancer with frequent amplification of MYCN. Genome-wide expression analysis showed downregulation of the MYCN transcriptional program accompanied by suppression of MYCN transcription. Functionally, bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in neuroblastoma. BRD4 knockdown phenocopied these effects, establishing BET bromodomains as transcriptional regulators of MYCN. BET inhibition conferred a significant survival advantage in 3 in vivo neuroblastoma models, providing a compelling rationale for developing BET bro- modomain inhibitors in patients with neuroblastoma. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -
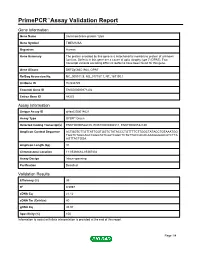
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name transmembrane protein 126A Gene Symbol TMEM126A Organism Human Gene Summary The protein encoded by this gene is a mitochondrial membrane protein of unknown function. Defects in this gene are a cause of optic atrophy type 7 (OPA7). Two transcript variants encoding different isoforms have been found for this gene. Gene Aliases DKFZp586C1924, OPA7 RefSeq Accession No. NC_000011.9, NG_017157.1, NT_167190.1 UniGene ID Hs.533725 Ensembl Gene ID ENSG00000171202 Entrez Gene ID 84233 Assay Information Unique Assay ID qHsaCID0014621 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000528105, ENST00000304511, ENST00000532180 Amplicon Context Sequence ACTGGTCTTGTTATTGGTGGTCTATACCCTGTTTTCTTGGCTATACCTGTAAATGG TGGTCTAGCAGCCAGGTATCAATCAGCTCTGTTACCACACAAAGGGAACATCTTA AGTTACTGGA Amplicon Length (bp) 91 Chromosome Location 11:85366682-85367402 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 99 R2 0.9997 cDNA Cq 21.12 cDNA Tm (Celsius) 80 gDNA Cq 35.93 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report TMEM126A, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Novel Likely Pathogenic Variants in TMEM126A Identified in Non
Kloth et al. BMC Medical Genetics (2019) 20:62 https://doi.org/10.1186/s12881-019-0795-x CASEREPORT Open Access Novel likely pathogenic variants in TMEM126A identified in non-syndromic autosomal recessive optic atrophy: two case reports Katja Kloth1, Matthis Synofzik2,3, Christoph Kernstock4, Simone Schimpf-Linzenbold5, Frank Schuettauf6, Axel Neu7, Bernd Wissinger4 and Nicole Weisschuh4* Abstract Background: Reports on autosomal recessive optic atrophy (arOA) are sparse and so far, only one gene has been specifically associated with non-syndromic arOA, namely TMEM126A. To date, all reports of pathogenic TMEM126A variants are from affected individuals of Maghrebian origin, who all carry an identical nonsense variant. Here we report two novel variants in the TMEM126A gene from non-Maghreb individuals, both found in affected individuals with an arOA phenotype. Case presentation: We report three affected individuals from two families. The proband of family A, a 24-year-old Turkish woman, was diagnosed with visual loss in early childhood but a diagnosis of optic atrophy was only made at 14 years. A diagnostic gene panel revealed a splice donor variant (c.86 + 2 T > C) in homozygous state in the TMEM126A gene. Analysis of this variant based on RNA from whole blood revealed a single aberrant transcript lacking exon 2, presumably representing a functional null allele. Two siblings from family B, a 16-year old Iraqi girl and her 14-year old brother, were diagnosed with optic atrophy in early childhood. A missense variant p.(S36 L) in the TMEM126A gene was identified in homozygous state in a gene panel-based diagnostic setting in both siblings. -

Molecular Characterization of Mutant Mouse Strains Generated from the EUCOMM/KOMP-CSD ES Cell Resource
Mamm Genome DOI 10.1007/s00335-013-9467-x Molecular Characterization of Mutant Mouse Strains Generated from the EUCOMM/KOMP-CSD ES Cell Resource Edward Ryder • Diane Gleeson • Debarati Sethi • Sapna Vyas • Evelina Miklejewska • Priya Dalvi • Bishoy Habib • Ross Cook • Matthew Hardy • Kalpesh Jhaveri • Joanna Bottomley • Hannah Wardle-Jones • James N. Bussell • Richard Houghton • Jennifer Salisbury • William C. Skarnes • Sanger Mouse Genetics Project • Ramiro Ramirez-Solis Received: 3 April 2013 / Accepted: 27 June 2013 Ó The Author(s) 2013. This article is published with open access at Springerlink.com Abstract The Sanger Mouse Genetics Project generates Introduction knockout mice strains using the EUCOMM/KOMP-CSD embryonic stem (ES) cell collection and characterizes the The extensive genetic resources available for the mouse, consequences of the mutations using a high-throughput including the sequencing and annotation of the genomes of primary phenotyping screen. Upon achieving germline multiple inbred laboratory strains (Church et al. 2009; transmission, new strains are subject to a panel of quality Keane et al. 2011; Flicek et al. 2012; Wong et al. 2012), control (QC) PCR- and qPCR-based assays to confirm the have facilitated comprehensive comparisons with the correct targeting, cassette structure, and the presence of the human genome (Guigo et al. 2003; Zheng-Bradley et al. 30 LoxP site (required for the potential conditionality of the 2010; Mouse ENCODE Consortium et al. 2012). This allele). We report that over 86 % of the 731 strains studied makes the mouse a powerful tool for both investigating showed the correct targeting and cassette structure, of gene function and modelling disease progression in mam- which 97 % retained the 30 LoxP site. -

Gnomad Lof Supplement
1 gnomAD supplement gnomAD supplement 1 Data processing 4 Alignment and read processing 4 Variant Calling 4 Coverage information 5 Data processing 5 Sample QC 7 Hard filters 7 Supplementary Table 1 | Sample counts before and after hard and release filters 8 Supplementary Table 2 | Counts by data type and hard filter 9 Platform imputation for exomes 9 Supplementary Table 3 | Exome platform assignments 10 Supplementary Table 4 | Confusion matrix for exome samples with Known platform labels 11 Relatedness filters 11 Supplementary Table 5 | Pair counts by degree of relatedness 12 Supplementary Table 6 | Sample counts by relatedness status 13 Population and subpopulation inference 13 Supplementary Figure 1 | Continental ancestry principal components. 14 Supplementary Table 7 | Population and subpopulation counts 16 Population- and platform-specific filters 16 Supplementary Table 8 | Summary of outliers per population and platform grouping 17 Finalizing samples in the gnomAD v2.1 release 18 Supplementary Table 9 | Sample counts by filtering stage 18 Supplementary Table 10 | Sample counts for genomes and exomes in gnomAD subsets 19 Variant QC 20 Hard filters 20 Random Forest model 20 Features 21 Supplementary Table 11 | Features used in final random forest model 21 Training 22 Supplementary Table 12 | Random forest training examples 22 Evaluation and threshold selection 22 Final variant counts 24 Supplementary Table 13 | Variant counts by filtering status 25 Comparison of whole-exome and whole-genome coverage in coding regions 25 Variant annotation 30 Frequency and context annotation 30 2 Functional annotation 31 Supplementary Table 14 | Variants observed by category in 125,748 exomes 32 Supplementary Figure 5 | Percent observed by methylation. -

Determining the Causes of Recessive Retinal Dystrophy
Determining the causes of recessive retinal dystrophy By Mohammed El-Sayed Mohammed El-Asrag BSc (hons), MSc (hons) Genetics and Molecular Biology Submitted in accordance with the requirements for the degree of Doctor of Philosophy The University of Leeds School of Medicine September 2016 The candidate confirms that the work submitted is his own, except where work which has formed part of jointly- authored publications has been included. The contribution of the candidate and the other authors to this work has been explicitly indicated overleaf. The candidate confirms that appropriate credit has been given within the thesis where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Mohammed El-Sayed Mohammed El-Asrag to be identified as author of this work has been asserted by his in accordance with the Copyright, Designs and Patents Act 1988. © 2016 The University of Leeds and Mohammed El-Sayed Mohammed El-Asrag Jointly authored publications statement Chapter 3 (first results chapter) of this thesis is entirely the work of the author and appears in: Watson CM*, El-Asrag ME*, Parry DA, Morgan JE, Logan CV, Carr IM, Sheridan E, Charlton R, Johnson CA, Taylor G, Toomes C, McKibbin M, Inglehearn CF and Ali M (2014). Mutation screening of retinal dystrophy patients by targeted capture from tagged pooled DNAs and next generation sequencing. PLoS One 9(8): e104281. *Equal first- authors. Shevach E, Ali M, Mizrahi-Meissonnier L, McKibbin M, El-Asrag ME, Watson CM, Inglehearn CF, Ben-Yosef T, Blumenfeld A, Jalas C, Banin E and Sharon D (2015). -

Type 2 Diabetes Modifies Skeletal Muscle Gene Expression Response to Gastric Bypass Surgery
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.19.427352; this version posted January 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Type 2 Diabetes Modifies Skeletal Muscle Gene Expression Response to Gastric Bypass Surgery Matthew D. Barberio1,2, G. Lynis Dohm3, Walter J. Pories4, Natalie A. Gadaleta2, Joseph A. Houmard5, Evan P. Nadler6, Monica J. Hubal1,7 1Center for Genetic Medicine Research, Children’s National Medical Center, Washington, D.C. 2Department of Exercise and Nutrition Sciences, Milken Institute School of Public Health, George Washington University, Washington, D.C. 3Department of Physiology, Brody School of Medicine, East Carolina University, Greenville, N.C. 4Department of Surgery, Brody School of Medicine, East Carolina University, Greenville, N.C. 5Human Performance Laboratory, Department of Kinesiology, College of Health and Human Performance, East Carolina University, Greenville, N.C. 6Division of Pediatric Surgery, Children’s National Medical Center, Washington, D.C. 7 Department of Kinesiology, Indiana University Purdue University Indianapolis, Indianapolis, IN Running Title: Skeletal Muscle Gene Expression Following Weight-Loss Surgery Word Count: 3325 Tables: 3 Figures: 3 Corresponding Author: Monica J. Hubal, PhD Associate Professor, Dept. of Kinesiology Indiana University Purdue University – Indianapolis 901 West New York St.; PE266 Indianapolis, IN 46202-5192 Email: [email protected] Telephone: 317-278-2343 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.19.427352; this version posted January 20, 2021. -

Loss of TMEM126A Promotes Extracellular Matrix Remodeling
Cancer Letters 440–441 (2019) 189–201 Contents lists available at ScienceDirect Cancer Letters journal homepage: www.elsevier.com/locate/canlet Original Articles Loss of TMEM126A promotes extracellular matrix remodeling, epithelial-to- T mesenchymal transition, and breast cancer metastasis by regulating mitochondrial retrograde signaling ∗∗ He-Fen Suna,b, , Xue-li Yanga,b, Yang Zhaoa,b, Qi Tianc, Meng-Ting Chena,b, Yuan-yuan Zhaoa,b, ∗ Wei Jina,b, a Department of Breast Surgery, Key Laboratory of Breast Cancer in Shanghai, Fudan University Shanghai Cancer Center, Shanghai, 200030, China b Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, 200030, China c Department of Obstetrics and Gynecology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, China ARTICLE INFO ABSTRACT Keywords: TMEM126A is a mitochondrial transmembrane protein, and its functions in breast cancer progression remain TMEM126A unclear. In this study, via the iTRAQ assay using primary and metastatic breast cancer cell models, we found that Metastasis TMEM126A expression decreased in metastatic cells. We further confirmed that low TMEM126A expression ECM correlated with tumor progression and poor prognosis in patients. The downregulation of TMEM126A in breast ROS cancer cell lines significantly enhanced the metastatic properties in vitro and in vivo, whereas its overexpression decreased the metastatic potential of cell lines. Mechanistic studies based on RNA-sequencing indicated that TMEM126A might regulate cell metastasis via ECM-receptor interaction, focal adhesions, and actin cytoskeleton, among other processes. Furthermore, the loss of TMEM126A activated extracellular matrix (ECM) remodeling and promoted epithelial-to-mesenchymal transition (EMT). Moreover, TMEM126A silencing induced reactive oxygen species (ROS) production and mitochondrial membrane potential depolarization. -

Genetic Testing for Developmental Disabilities, Intellectual Disability, and Autism Spectrum Disorder Technical Brief Number 23
Technical Brief Number 23 Genetic Testing for Developmental Disabilities, Intellectual Disability, and Autism Spectrum Disorder Technical Brief Number 23 Genetic Testing for Developmental Disabilities, Intellectual Disability, and Autism Spectrum Disorder Prepared for: Agency for Healthcare Research and Quality U.S. Department of Health and Human Services 540 Gaither Road Rockville, MD 20850 www.ahrq.gov Contract No. 290-2012-00011-I Prepared by: ECRI Institute–Penn Medicine Evidence-based Practice Center Plymouth Meeting, PA Investigators: Fang Sun, M.D., Ph.D. Jeff Oristaglio, Ph.D. Susan E. Levy, M.D., M.P.H. Hakon Hakonarson, M.D., Ph.D. Nancy Sullivan, B.A. Joann Fontanarosa, Ph.D. Karen M. Schoelles, M.D., M.S., FACP AHRQ Publication No. 15-EHC024-EF June 2015 This report is based on research conducted by the ECRI–Penn Medicine AHRQ Evidence-based Practice Center (EPC) under contract to the Agency for Healthcare Research and Quality (AHRQ), Rockville, MD (290-2012-00011-I). The findings and conclusions in this document are those of the authors, who are responsible for its contents; the findings and conclusions do not necessarily represent the views of AHRQ. Therefore, no statement in this report should be construed as an official position of AHRQ or of the U.S. Department of Health and Human Services. The information in this report is intended to help health care decisionmakers—patients and clinicians, health system leaders, and policymakers, among others—make well-informed decisions and thereby improve the quality of health care services. This report is not intended to be a substitute for the application of clinical judgment. -

Next-Generation Sequencing in the Diagnosis of Dementia And
Next‐generation sequencing in the diagnosis of Dementia and Huntington’s disease Phenocopy Syndromes A thesis submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy from University College London Carolin Anna Maria Koriath Institute of Neurology Department of Molecular Neurodegeneration University College London 2020 1 2 Declaration I, Carolin Anna Maria Koriath, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. However, contemporary, large‐scale projects require a complex set of skills and extensive work best provided by a team. Like many studies nowadays, the investigation and analysis of the MRC Dementia Gene Panel study were based on a collaborative effort and contributions from different members of the Human Genetics Programme at the MRC Prion Institute at UCL. I have personally performed a significant amount of the laboratory work and will highlight these contributions in the methods section; I have also performed all stages of data processing. Namely, I have analysed all next‐generation sequencing data and developed criteria for the correct classification of identified variants. For time and cost advantages, samples for the Huntington’s disease phenocopy study were whole‐genome sequenced and aligned at Edinburgh Genomics, but I performed the data analysis. I have put this data into context with the clinical data for further analysis and the statistical workup, and, in close dialogue with my supervisors Prof Simon Mead and Prof Sarah Tabrizi, have drawn conclusions from all available data which are set out in this PhD thesis.