Transcriptomic Signature in Lymphoblastoid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 RESEARCH ARTICLE Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition Alexandre Puissant1,3, Stacey M. Frumm1,3, Gabriela Alexe1,3,5,6, Christopher F. Bassil1,3, Jun Qi2, Yvan H. Chanthery8, Erin A. Nekritz8, Rhamy Zeid2, William Clay Gustafson8, Patricia Greninger7, Matthew J. Garnett10, Ultan McDermott10, Cyril H. Benes7, Andrew L. Kung1,3, William A. Weiss8,9, James E. Bradner2,4, and Kimberly Stegmaier1,3,6 Downloaded from cancerdiscovery.aacrjournals.org on October 2, 2021. © 2013 American Association for Cancer Research. 15-CD-12-0418_p308-323.indd 1 22/02/13 12:15 AM Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 A BSTRACT Bromodomain inhibition comprises a promising therapeutic strategy in cancer, particularly for hematologic malignancies. To date, however, genomic biomarkers to direct clinical translation have been lacking. We conducted a cell-based screen of genetically defined cancer cell lines using a prototypical inhibitor of BET bromodomains. Integration of genetic features with chemosensitivity data revealed a robust correlation between MYCN amplification and sensitivity to bromodomain inhibition. We characterized the mechanistic and translational significance of this finding in neuroblastoma, a childhood cancer with frequent amplification of MYCN. Genome-wide expression analysis showed downregulation of the MYCN transcriptional program accompanied by suppression of MYCN transcription. Functionally, bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in neuroblastoma. BRD4 knockdown phenocopied these effects, establishing BET bromodomains as transcriptional regulators of MYCN. BET inhibition conferred a significant survival advantage in 3 in vivo neuroblastoma models, providing a compelling rationale for developing BET bro- modomain inhibitors in patients with neuroblastoma. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -
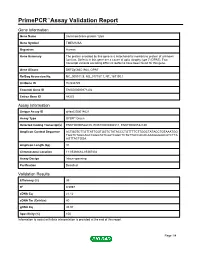
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name transmembrane protein 126A Gene Symbol TMEM126A Organism Human Gene Summary The protein encoded by this gene is a mitochondrial membrane protein of unknown function. Defects in this gene are a cause of optic atrophy type 7 (OPA7). Two transcript variants encoding different isoforms have been found for this gene. Gene Aliases DKFZp586C1924, OPA7 RefSeq Accession No. NC_000011.9, NG_017157.1, NT_167190.1 UniGene ID Hs.533725 Ensembl Gene ID ENSG00000171202 Entrez Gene ID 84233 Assay Information Unique Assay ID qHsaCID0014621 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000528105, ENST00000304511, ENST00000532180 Amplicon Context Sequence ACTGGTCTTGTTATTGGTGGTCTATACCCTGTTTTCTTGGCTATACCTGTAAATGG TGGTCTAGCAGCCAGGTATCAATCAGCTCTGTTACCACACAAAGGGAACATCTTA AGTTACTGGA Amplicon Length (bp) 91 Chromosome Location 11:85366682-85367402 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 99 R2 0.9997 cDNA Cq 21.12 cDNA Tm (Celsius) 80 gDNA Cq 35.93 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report TMEM126A, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

The DNA Sequence and Comparative Analysis of Human Chromosome 20
articles The DNA sequence and comparative analysis of human chromosome 20 P. Deloukas, L. H. Matthews, J. Ashurst, J. Burton, J. G. R. Gilbert, M. Jones, G. Stavrides, J. P. Almeida, A. K. Babbage, C. L. Bagguley, J. Bailey, K. F. Barlow, K. N. Bates, L. M. Beard, D. M. Beare, O. P. Beasley, C. P. Bird, S. E. Blakey, A. M. Bridgeman, A. J. Brown, D. Buck, W. Burrill, A. P. Butler, C. Carder, N. P. Carter, J. C. Chapman, M. Clamp, G. Clark, L. N. Clark, S. Y. Clark, C. M. Clee, S. Clegg, V. E. Cobley, R. E. Collier, R. Connor, N. R. Corby, A. Coulson, G. J. Coville, R. Deadman, P. Dhami, M. Dunn, A. G. Ellington, J. A. Frankland, A. Fraser, L. French, P. Garner, D. V. Grafham, C. Grif®ths, M. N. D. Grif®ths, R. Gwilliam, R. E. Hall, S. Hammond, J. L. Harley, P. D. Heath, S. Ho, J. L. Holden, P. J. Howden, E. Huckle, A. R. Hunt, S. E. Hunt, K. Jekosch, C. M. Johnson, D. Johnson, M. P. Kay, A. M. Kimberley, A. King, A. Knights, G. K. Laird, S. Lawlor, M. H. Lehvaslaiho, M. Leversha, C. Lloyd, D. M. Lloyd, J. D. Lovell, V. L. Marsh, S. L. Martin, L. J. McConnachie, K. McLay, A. A. McMurray, S. Milne, D. Mistry, M. J. F. Moore, J. C. Mullikin, T. Nickerson, K. Oliver, A. Parker, R. Patel, T. A. V. Pearce, A. I. Peck, B. J. C. T. Phillimore, S. R. Prathalingam, R. W. Plumb, H. Ramsay, C. M. -

Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells
International Journal of Molecular Sciences Article Exosomes from Plasma of Neuroblastoma Patients Contain Doublestranded DNA Reflecting the Mutational Status of Parental Tumor Cells Chiara Degli Esposti 1,† , Barbara Iadarola 1,† , Simone Maestri 1 , Cristina Beltrami 1, Denise Lavezzari 1 , Martina Morini 2 , Patrizia De Marco 3, Giovanni Erminio 4 , Alberto Garaventa 5, Federico Zara 3, Massimo Delledonne 1 , Marzia Ognibene 3,* and Annalisa Pezzolo 6 1 Dipartimento di Biotecnologie, Università degli Studi di Verona, 37134 Verona, Italy; [email protected] (C.D.E.); [email protected] (B.I.); [email protected] (S.M.); [email protected] (C.B.); [email protected] (D.L.); [email protected] (M.D.) 2 Laboratorio di Biologia Molecolare, IRCCS Giannina Gaslini, 16147 Genova, Italy; [email protected] 3 U.O.C. Genetica Medica, IRCCS Giannina Gaslini, 16147 Genova, Italy; [email protected] (P.D.M.); [email protected] (F.Z.) 4 Epidemiologia e Biostatistica, IRCCS Giannina Gaslini, 16147 Genova, Italy; [email protected] 5 Divisione di Oncologia, IRCCS Giannina Gaslini, 16147 Genova, Italy; [email protected] 6 IRCCS Giannina Gaslini, 16147 Genova, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-010-56362601 † These authors contributed equally to this work. Citation: Degli Esposti, C.; Iadarola, Abstract: Neuroblastoma (NB) is an aggressive infancy tumor, leading cause of death among B.; Maestri, S.; Beltrami, C.; Lavezzari, preschool age diseases. Here we focused on characterization of exosomal DNA (exo-DNA) isolated D.; Morini, M.; De Marco, P.; Erminio, from plasma cell-derived exosomes of neuroblastoma patients, and its potential use for detection of G.; Garaventa, A.; Zara, F.; et al. -

Identification of Potential Genes and Mirnas Associated with Sepsis Based on Microarray Analysis
MOLECULAR MEDICINE REPORTS 17: 6227-6234, 2018 Identification of potential genes and miRNAs associated with sepsis based on microarray analysis YIN LI*, FENGXIA ZHANG*, YAN CONG and YUN ZHAO Emergency Department, Huadong Hospital, Shanghai 200040, P.R. China Received May 10, 2017; Accepted September 18, 2017 DOI: 10.3892/mmr.2018.8668 Abstract. Sepsis is a type of systemic inflammatory response the MCODE module. In the integrated DEG‑miRNA‑TF regu- syndrome caused by infection. The present study aimed to latory network, hsa‑miR‑150 (target gene MYLK3) and 21 TFs, examine key genes and microRNAs (miRNAs) involved in comprising 14 upregulated DEGs (including MYC) and seven the pathogenesis of sepsis. The GSE13205 microarray dataset, downregulated DEGs, were identified. The results suggested downloaded from the Gene Expression Omnibus was analyzed that NOP14, NOP2, AATF, GTPBP4, BYSL, MYC, MYLK3 and using bioinformatics tools, and included muscle biopsy speci- miR‑150 may be involved in the pathogenesis of sepsis. mens of 13 patients with sepsis and eight healthy controls. The differentially expressed genes (DEGs) in samples from Introduction patients with sepsis were identified using the Linear Models for Microarray package in R language. Using the Database Sepsis, a type of systemic inflammatory response syndrome for Annotation, Visualization and Integration Discovery (SIRS), is a complication caused by the response of the whole tool, functional and pathway enrichment analyses were body, and injured tissues and organs to an infection (1). In the performed to examine the potential functions of the DEGs. last decade, the incidence rates of sepsis and severe sepsis in The protein‑protein interaction (PPI) network was constructed the population were 4.37 and 2.70 cases per 1,000 individuals, with the DEGs using the Search Tool for the Retrieval of respectively, in high‑income‑countries (including seven Interacting Genes, and the network topology was analyzed countries on four continents); annually, the global mortality using CytoNCA. -

GTPBP4 (T-15): Sc-160426
SAN TA C RUZ BI OTEC HNOL OG Y, INC . GTPBP4 (T-15): sc-160426 BACKGROUND PRODUCT Small G proteins act as molecular switches for regulation of variety of cellu lar Each vial contains 200 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide processes, such as nuclear transport, signal transduction, membrane traffick - and 0.1% gelatin. ing and protein synthesis. GTPBP4 (GTP binding protein 4), also designated Blocking peptide available for competition studies, sc-160426 P, (100 µg nucleolar GTP-binding protein 1, chronic renal failure gene protein (CRFG), pep tide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). NOG1 or NGB, is a 634 amino acid novel GTP-binding protein that plays a role in 60S ribosomal subunit biogenesis and belongs to the GTP1/OBG family and APPLICATIONS NOG subfamily. Localizing to nucleolus, GTPBP4 has been observed to inhibit cell aggregation and growth when ectopically expressed in tumorigenic GTPBP4 (T-15) is recommended for detection of GTPBP4 of mouse, rat and schwanomma cells, and is believed to act as a tumor supressor when work ing human origin by Western Blotting (starting dilution 1:200, dilution range in conjunction with NF2 (merlin). GTPBP4 contains multiple phosphorylated 1:100-1:1000), immunofluorescence (starting dilution 1:50, dilution range amino acid residues and is encoded by a gene located on human chromo some 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution range 10p15.3. 1:30- 1:3000); non cross-reactive with other GTPBP family members. -

SUPPLEMENTARY INFORMATION APPENDIX the Draft
SUPPLEMENTARY INFORMATION APPENDIX The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau Xingquan Zenga,b,1, Hai Longc,1, Zhuo Wangd,1, Shancen Zhaod,1, Yawei Tanga,b,1, Zhiyong Huangd,1, Yulin Wanga,b,1, Qijun Xua,b, Likai Maod, Guangbing Dengc, Xiaoming Yaod, Xiangfeng Lid,e, Lijun Baid, Hongjun Yuana,b, Zhifen Panc, Renjian Liua,b, Xin Chenc, QiMei WangMua,b, Ming Chend, Lili Yud, Junjun Liangc, DaWa DunZhua,b, Yuan Zhengd, Shuiyang Yuc, ZhaXi LuoBua,b, Xuanmin Guangd, Jiang Lid, Cao Dengd, Wushu Hud, Chunhai Chend, XiongNu TaBaa,b, Liyun Gaoa,b, Xiaodan Lvd, Yuval Ben Abuf, Xiaodong Fangd, Eviatar Nevog,2, Maoqun Yuc,2, Jun Wangh,i,j,2, Nyima Tashia,b,2 aTibet Academy of Agricultural and Animal Husbandry Sciences, Lhasa, Tibet 850002, China; bBarley Improvement and Yak Breeding Key Laboratory of Tibet Autonomous Region, Lhasa 850002, China; cChengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, P. R. China; dBGI-Tech, BGI-Shenzhen, Shenzhen 518083, China; eCollege of Life Science, University of Chinese Academy of Sciences, Beijing 100049, China; fProjects and Physics Section, Sapir Academic College, D.N. Hof Ashkelon 79165, Israel; gInstitute of Evolution, University of Haifa, Mount Carmel, Haifa 31905, Israel; hBGI-Shenzhen, Shenzhen 518083, China; iDepartment of Biology, University of Copenhagen, Copenhagen 2200, Denmark; jPrincess Al Jawhara Center of Excellence in the Research of Hereditary Disorders, King Abdulaziz University, Jeddah 21441, Saudi Arabia 1These authors contributed equally to this work. 2To whom correspondence should be addressed. Email: Nyima Tashi, [email protected]; Jun Wang, [email protected]; Maoqun Yu, [email protected]; or Eviatar Nevo, [email protected]. -

Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’S Disease
SAGE-Hindawi Access to Research International Journal of Alzheimer’s Disease Volume 2010, Article ID 604792, 56 pages doi:10.4061/2010/604792 Research Article Complex and Multidimensional Lipid Raft Alterations in a Murine Model of Alzheimer’s Disease Wayne Chadwick, 1 Randall Brenneman,1, 2 Bronwen Martin,3 and Stuart Maudsley1 1 Receptor Pharmacology Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA 2 Miller School of Medicine, University of Miami, Miami, FL 33124, USA 3 Metabolism Unit, National Institute on Aging, National Institutes of Health, 251 Bayview Boulevard, Suite 100, Baltimore, MD 21224, USA Correspondence should be addressed to Stuart Maudsley, [email protected] Received 17 May 2010; Accepted 27 July 2010 Academic Editor: Gemma Casadesus Copyright © 2010 Wayne Chadwick et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Various animal models of Alzheimer’s disease (AD) have been created to assist our appreciation of AD pathophysiology, as well as aid development of novel therapeutic strategies. Despite the discovery of mutated proteins that predict the development of AD, there are likely to be many other proteins also involved in this disorder. Complex physiological processes are mediated by coherent interactions of clusters of functionally related proteins. Synaptic dysfunction is one of the hallmarks of AD. Synaptic proteins are organized into multiprotein complexes in high-density membrane structures, known as lipid rafts. These microdomains enable coherent clustering of synergistic signaling proteins.